Download

1 / 31
310 likes | 670 Views
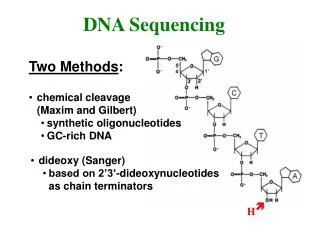
DNA sequencing. Dideoxy analogs of normal nucleotide triphosphates (ddNTP) cause premature termination of a growing chain of nucleotides. ACAGTCGATTG ACAddG ACAGTCddG ACAGTCGATTddG

E N D
DNA sequencing. • Dideoxy analogs of normal nucleotide triphosphates (ddNTP) cause premature termination of a growing chain of nucleotides. ACAGTCGATTG ACAddG ACAGTCddG ACAGTCGATTddG • Fragments are separated according to their sizes in gel electrophoresis. The lengths show the positions of “G” in the original DNA sequence.
Nucleotides and phosphodiester bond. Phosphodiester bond
Genomic sequencing. • Individual chromosomes are broken into 100kb random fragments. • This library of fragments is screened to find overlapping fragments – contigs. • Unique overlapping clones are chosen for sequencing. • Put together overlapping sequenced clones using computer programs.
Sequencing cDNA libraries. • mRNA is pooled from the tissues which express genes. • cDNA libraries are prepared by copying of mRNA with reverse transcriptase. • Expressed Sequence Tags (EST) – partial sequences of expressed genes. • Comparing translated ESTs to annotated proteins – annotation of genes.
Gene prediction. Gene – DNA sequence encoding protein, rRNA, tRNA … Gene concept is complicated: • Introns/exons • Alternative splicing • Genes-in-genes • Multisubunit proteins
Gene structure. ATG TER -35 -10 Promoter sequences Gene ATG – start codon; TER (TAA, TAG,TGA) – termination codons
Codon usage tables. • Each amino acid can be encoded by several codons. • Each organism has characteristic pattern of codon usage.
Problems arising in gene prediction. • Distinguishing pseudogenes (not working former genes) from genes. • Exon/intron structure in eukaryotes, exon flanking regions – not very well conserved. • Exon can be shuffled alternatively – alternative splicing. • Genes can overlap each other and occur on different strands of DNA.
Gene identification • Homology-based gene prediction • Similarity Searches (e.g. BLAST, BLAT) • ESTs • Ab initio gene prediction • Prokaryotes • ORF identification • Eukaryotes • Promoter prediction • PolyA-signal prediction • Splice site, start/stop-codon predictions
Ab initio gene prediction. Predictions are based on the observation that gene DNA sequence is not random: - Gene-coding sequence has start and stop codons. • Each species has a characteristic pattern of synonymous codon usage. • Non-coding ORFs are very short. • Gene would correspond to the longest ORF. These methods look for the characteristic features of genes and score them high.
Prokaryotic genes – searching for ORFs. • Small genomes have high gene density Haemophilus influenza – 85% genic • No introns • Operons One transcript, many genes • Open reading frames (ORF) – contiguous set of codons, start with Met-codon, ends with stop codon.
Example of ORFs. There are six possible ORFs in each sequence for both directions of transcription.
Gene preference score – important indicator of coding region. Observation: frequencies of codons and codon pairs in coding and non-coding regions are different. Given a sequence of codons: and assuming independence, the probability of finding coding region: The probability of finding sequence “C” in non-coding regions: The gene preference score:
Classwork I. Calculate the gene preference score for the following human DNA sequence: AGTACA
Ab initio gene prediction methods. • Grail II – predicts exons, promoters, Poly(A) sites. Neural network plus dynamic programming. • GeneParser – predicts the most likely combination of exons/introns. Dynamic programming. • GeneMark – mostly for prokaryotes, Hidden Markov Models. • GeneScan – Fourier transform of DNA sequence to find characteristic patterns.
Confirming gene location using EST libraries. • Expressed Sequence Tags (ESTs) – sequenced short segments of cDNA. They are organized in the database “UniGene”. • If region matches ESTs with high statistical significance, then it is a gene or pseudogene.
Gene prediction accuracy. True positives (TP) – nucleotides, which are correctly predicted to be within the gene. Actual positives (AP) – nucleotides, which are located within the actual gene. Predicted positives (PP) – nucleotides, which are predicted in the gene. Sensitivity = TP / AP Specificity = TP / PP
Gene prediction accuracy. GenScan Website
Common difficulties • First and last exons difficult to annotate because they contain UTRs. • Smaller genes are not statistically significant so they are thrown out. • Algorithms are trained with sequences from known genes which biases them against genes about which nothing is known.
Gene prediction: classwork II. • Go to http://www.ncbi.nlm.nih.gov/mapview/ and view all hemoglobin genes of H. sapiens • Find 6 hemoglobin genes on chromosome 11, view the DNA sequence of this chromosome region • Submit this sequence to GenScan server at http://genes.mit.edu/GENSCAN.html
Genome analysis. Genome – the sum of genes and intergenic sequences of haploid cell.
The value of genome sequences lies in their annotation • Annotation – Characterizing genomic features using computational and experimental methods • Genes: levels of annotation • Gene Prediction – Where are genes? • What do they encode? • What proteins/pathways involved in?
Accuracy of genome annotation. • In most genomes functional predictions has been made for majority of genes 54-79%. • The source of errors in annotation: - overprediction (those hits which are statistically significant in the database search are not checked) - multidomain protein (found the similarity to only one domain, although the annotation is extended to the whole protein). The error of the genome annotation can be as big as 25%.
Sample genomes. • There is almost no correlation between the number of genes and organism’s complexity. • There is a correlation between the amount of nonprotein-coding DNA and complexity.
Comparative genomics - comparison of gene number, gene content and gene location in genomes.. Campbell & Heyer “Genomics”
Analysis of gene order (synteny). Genes with a related function are frequently clustered on the chromosome. Ex: E.coli genes responsible for synthesis of Trp are clustered and order is conserved between different bacterial species. Operon: set of genes transcribed simultaneously with the same direction of transcription
Analysis of gene order (synteny). Koonin & Galperin “Sequence, Evolution, Function”
Analysis of gene order (synteny). • The order of genes is not very well conserved if %identity between prokaryotic genomes is < 50% • The gene neighborhood can be conserved so that the all neighboring genes belong to the same functional class. • Functional prediction based on gene neighboring.
Classwork III: Comparing microbial genomes. • Go to http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi • Select Thermus thermophilus genome • View TaxTable • What gene clusters do you see which are common with Archaea?