Download

1 / 16
160 likes | 261 Views
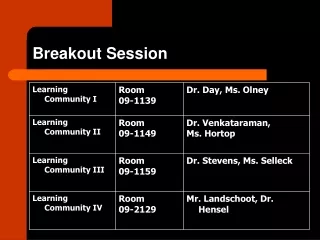
Best Practices and Application of GMPs for Small Molecule Drugs in Early Development IQ Workshop, Feb 4-5, 2014, Washington, D.C. Specifications Breakout Session 2 Pete Yehl and Mike Coutant , moderators. Best Practices and GMPs in Early Development Problem Statement.

E N D
Best Practices and Application of GMPs for Small Molecule Drugs in Early Development IQ Workshop, Feb 4-5, 2014, Washington, D.C. Specifications Breakout Session 2 Pete Yehl and Mike Coutant, moderators
Best Practices and GMPs in Early DevelopmentProblem Statement Improved clarity in definition of GMP expectations for early development (Phase I to 2a) would advance innovation in drug product development Potential to improve cycle times and reduce costs, while maintaining appropriate product quality and always ensuring patient safety Each company in Industry interprets existing GMP guidances according to its own culture and risk tolerance In part, this is due to the vague nature of the guidances available regarding GMP expectations in early dev (e.g. Q7A, Phase 1 GMPs, etc) Internal Industry debates between QA and CMC groups often result in conservative “one-size fits all” interpretations across the dev continuum Feedback from the previous 2007 PhRMA initiative: Biggest roadblock to achieving greater flexibility in early development is “ourselves” in Industry A common approach to best practices and GMP expectations for early development is needed to build alignment with regulatory agencies and internal stakeholders
Highlights of the IQ Specifications Paper Scope Small molecules (biologics not included) Relevant to solid oral dosage forms US Regulatory filings (concepts should be applicable to rest of world) General Concepts ICH Q6A appropriate for commercial not Early Development Commercial specifications should be tied to final process validation results Process, formulation, and method changes expected in early Dev so Specs evolve During late development, maturing product and process understanding Risk based approach: Early clinical has safety focus ⇒ wider acceptance criteria (AC) High attrition rate for compounds in early development Late development growing product and process understanding ⇒ maturing specs Differentiating Early Phase Spec Testing by standardizing typical tests and AC Release (submitted in regulatory filings) Stability (to establish DS and DP retest dates) Internal (for informational purposes only, but can have tighter AC) • IQ Specs paper intended to be provocative starting point to stimulate discussion 3
Topic 1: General Control Strategies IQ proposal: In-Process control vs. Internal Target vs. Regulatory Specification In addition to release and stability tests, consideration is given to internal tests and acceptance criteria that are not normally part of formal specifications. These internal tests can be performed to collect information for product and process understanding, or to allow for tighter control (i.e., target criteria tighter than the release testing criteria) to ensure product quality will be maintained throughout the product’s retest period. Based on the information obtained in early development, additional tests and acceptance criteria for other attributes (e.g. water content) can be included as the late development focus shifts to process and product performance and consistency; eventually aligning the available ICH guidelines. Discussion: Use of IPCs vs. formal internal targets vs. registered specifications throughout development When does something move from being an internal target to registered specification? For PIC/PIB formulations, can purity be assigned through API testing? • Agreement on the general use of internal targets as a means of impurity control in Early Development. Understanding when it is and isn’t acceptable to monitor internally vs. having a formal regulatory specification 4
Topic 1: General Control Strategies IQ proposal: Use of IPCs to control chemical, chiral, solvent and residual metal/inorganic impurities in final API and Drug product DP intermediates (i.e. spray dry, HME) control of solvents and degradation products Consider intermediate testing for residual solvents, chiral purity Methods in place to control chemical impurities in API through RSM control Acceptability of using internal tests to obviate the need to submit additional test data and methods for health authority review would be contingent on product and materials knowledge level Balance of upfront effort to qualify IPC or intermediate test methods in lieu of testing API and validation would be a business decision Discussion: Use of IPCs vs. formal internal targets vs. registered API specifications and methods through various development stages Could this be applied DP intermediates? Early QBD approach to development – is this a business risk that companies would consider? Would internal testing and control of intermediates be sufficient? • Agreement on the general use of internal targets as a means of impurity control in Early Development. Understanding when it is and isn’t acceptable to monitor internally vs. having a formal regulatory specification 5
Topic 2: Impurity Control Strategies IQ proposal: 3x ICH limits for ID and qualification for Phase I and Phase II, transition to ICH specifications for pivotal studies Staged approach aligns with other industry initiatives (i.e., staged TTC PGI’s) Subject exposure lower and study durations are shorter. Subject population closely monitored IQ Proposal: Specification of synthetic process related impurities only in API, not in DP Discussion: Qualification threshold for early development ID vs. commercial/pre-commercial Does a staged approach have merit based on exposure arguments? Do internal impurity qualification thresholds differ from filed specifications? Any special considerations given for oncology drugs? General acceptance of process related impurity specifications limited to API unless they grow or change in the DP? • Agreement on development stage/phase based approach to impurity control and specifications • based on overall patient exposure and clinical care 6
Single set of tests and acceptance criteria (AC) for Phase 1 and 2a DS Specs IQ Specifications Paper: DS Summary 7
Single set of tests and AC for PIB/PIC formulations (rely primarily on DS results) IQ Specifications Paper: PIB/PIC DP Summary 8
IQ Specifications Paper: Capsule/Tablet Summary Single set of tests and AC for Phase 1 and 2a capsule/tablet Specs 9
Topic 3: Dissolution vs. Disintegration IQ proposal is that for rapidly dissolving immediate release formulations, it is recommended to include disintegration as a regulatory filed specification Dissolution may be performed as an internal specification (i.e., report results without defined acceptance criteria) to gather product knowledge during early development (e.g., for poorly soluble drugs). As additional knowledge is gained, dissolution acceptance criteria should be established in later development (i.e., Phase 2b and beyond). For rapidly dissolving immediate release formulations, it is recommended to include disintegration as a regulatory filed specification. Discussion: When is dissolution an appropriate registered specification? Have companies been filing disintegration successfully? Any country specific challenges? What API and DP properties or development milestones would trigger development of a dissolution method? • For Early Development, Disintegration is an appropriate drug product specification for rapidly dissolving immediate release formulations. Dissolution data may be generated to gather product knowledge. 10
Topic 4: Microbiological Testing IQ proposal is that micro testing on either API or DP is not required in early development for oral products. Manufacturing usually takes place in non-sterile facilities Since human GI is non-sterile, limited patient risk Testing sterility of API is business risk, even if used in parenteral Other bases for omitting micro testing on DP: Process water activity testing is an industry standard Capsule shells, other excipients are micro tested prior to use Discussion: What are the risks and strategies around microbiological testing for oral products in early development? Ancillary IV formulas produced using API made for oral use (for example in absolute BA studies) will still be micro tested (endotoxins and sterility) • An agreement that sterility testing for oral products is not needed in US in early development. A broader n industry perspective on experiences with other health authorities WRT micro testing • Especially in early development 11
Purpose and Publication Timing of the GMPs in Early Development Position Papers Intended as stimuli articles Serve as a starting point to stimulate further discussion at a future workshop Should not be perceived as final Industry position or recommendation Publication timing in Pharmaceutical Technology Overarching Summary – June 2012 Analytical Methods – July 2012 Drug Product Manufacturing – August 2012 Stability – September 2012 Specifications – Anticipated October 2012
IQ Workshop Objectives Provide a broader public forum for further discussion of the application of GMPs in Early Development as outlined in the position papers published in Pharmaceutical Technology in 2012 Build alignment with health authorities and industrial CMC stakeholders (QA, CMC development scientists, regulatory CMC) on the best practices outlined in the position papers. Discuss the impact of applying these concepts in early development for different therapeutic areas/disease states. Publish workshop proceedings integrating feedback based upon the discussions.
Best Practices and GMPs in Early Development: Scope and Strategy Limit initial scope to US filings to help build momentum Focus on Early Development (Phase I to 2a) Early = Supporting exploratory human clinical studies Late = Supporting pivotal human clinical studies Limited to small molecules (biologics excluded) Emphasis on 4 topics: Analytical method qualification/validation Drug product manufacturing Stability Specifications
GMPs in Early Development Part 5:Specifications To be published in October 2012 issue of Pharmaceutical Technology Volume 36, Issue 10, pp. 84-93 16