Download

1 / 14
140 likes | 478 Views
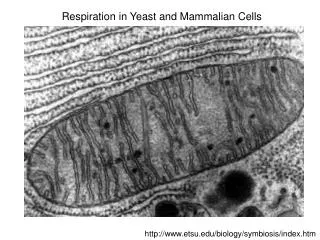
Mathematical modeling of protein aging and turnover in live yeast cells. Joseph Barry Huber Group, EMBL-Heidelberg 9 th Nov 2010. S. Cerevisiae. Protein localization Huh et al. “ Global analysis of protein localization in budding yeast”. Nature (2003) vol. 425 (6959) pp. 686- 691

E N D
Mathematical modeling of protein aging and turnover in live yeast cells Joseph Barry Huber Group, EMBL-Heidelberg 9th Nov 2010
S. Cerevisiae • Protein localization • Huh et al. “Global analysis of protein localization in budding yeast”. Nature (2003) vol. 425 (6959) pp. 686-691 • Protein half-lives • Belle et al. “Quantification of protein half-lives in the budding yeast proteome”. PNAS USA (2006) vol. 103 (35) pp. 13004-9 • Protein stability and local turnover only understood for certain proteins • e.g. Nuclear Pore Complex, see Khmelinskiiet al. (inreview), Knop Group, EMBL Protein stability and turnover in budding yeast (Image courtesy of Knop Group, EMBL) Aim of current research: To quantify stability and local turnover for all (~6000) protein species in S. Cerevisiae.
Snapshot Analysis Protein Stability (SAPS) PROTEIN mCherry sfGFP Dual fluorescence protein tag 43 min 5 min maturation half time* Using microscopy images for a single time point, age of protein may be determined by mathematical modeling Example fluorescence for NPC proteins* fluorescence intensity number of molecules *Khmelinskiiet al., “A quantitative imaging approach for the analysis of protein degradation and inheritance in living cells” (in review), Knop Group, EMBL-Heidelberg
Project Workflow Genome-wide library of yeast strains. One protein tagged for each strain. dual-reporter tagging Knop Group, EMBL-Heidelberg fluorescence microscopy Olympus scan^R image analysis mathematical modeling Bright Field mCherry sfGFP Huber Group, EMBL-Heidelberg Quantification of protein age and turnover
The case for mathematical modeling obtained from microscopy data mathematical model Protein populations Xi may be modeled as a function of relevant system parameters, for example: time maturation rates degradation rate protein production rate Fit mathematical model to experimental data to estimate values of system parameters.
Example mathematical model One-step GFP, two-step Cherry maturation mChe2 mChe1 mGFP CheON 1-GFPOFF CheON 2-GFPOFF p mGFP CheON 2-GFPON Dark CheOFF-GFPON CheON 1-GFPON mGFP mChe2 mChe1 all proteins degrade at same rate k Stochastic Model Deterministic Model ?
Deterministic Model (dashed lines) Stochastic Simulation (solid lines and shaded area) • Numerical solutions to rate equations solved using deSolve R package • Analytic solutions to rate equations also obtained • Implemented in C and called from R • Results averaged over 1,000 realisations Deterministic solution agrees with average from stochastic simulation Model used (stochastic or deterministic) will depend on protein population (small or large)
Example Movies Bright Field mCherry sfGFP • MCD1 promoter with dual-reporter construct • MCD1 protein: essential subunit of cohesin complex that helps hold sister chromatids together • Protein abundance peaks in S phase • Clear cell cycle dependence Special thanks to A. Kaufmann and S. Sasnouski
Can we model periodic signal of MCD1? Cell cycle dependent protein production rate Any periodic signal can be modeled as a Fourier Series: cell cycle time (approx 90 min) Fourier coefficients Is a periodic protein production rate sufficient to explain the periodic signal of MCD1 promotor?
Further modeling issues Cell division Occurs approximately every tcyl = 90 minutes Simple modeling approximation: 50% 50% (Future models will relax this condition) Fluorescence resonance energy transfer (FRET) Energy transfer possible from sfGFP to mCherry ONLY is the FRET efficiency mCherry sfGFP
Fitting experimental data to model Known parameters*: Fitting parameters: 6 parameter fit performed with R package “minpack.lm” *Khmelinskiiet al., “A quantitative imaging approach for the analysis of protein degradation and inheritance in living cells” (in review), Knop Group, EMBL-Heidelberg
Protein migration between compartments Compartment A Compartment B p OFF OFF a m m k k b ON ON Rate Equations: Steady-state and time-dependent solutions have been found. Next step: more accurate and complex migration models (stochastic and deterministic)
Outlook • Fitting of mathematical models to single time-point images. • Develop further models of between-compartment dynamics to determine local proteinturnover. • Explore more deeply the relationship between stochastic and deterministic models. • For further information on project, please see Andreas Kaufmann’s poster “Genome-wide analysis of protein stability of sub-cellular resolution”.
Acknowledgements Huber Group, EMBL-Heidelberg Wolfgang Huber Greg Pau Simon Anders Bernd Fischer Knop Group, EMBL-Heidelberg Michael Knop Andreas Kaufmann Anton Khmelinskii Sergey Sasnouski Matthias Meurer Special thanks to the EMBL retreat organization committee Thank you for listening!