Download

1 / 35
380 likes | 531 Views
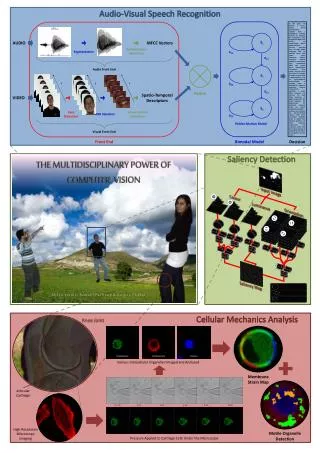
Proteomic analysis of cellular systems. molecular genetic lab 김윤식. Introduction. A prerequisite for a system-wide understanding of cellular processes is a precise knowledge of the principal actors.

E N D
Proteomic analysis of cellular systems molecular genetic lab 김윤식
Introduction • A prerequisite for a system-wide understanding of cellular processesis a precise knowledge of the principal actors. • The first such system-wide studies were performed at the level of mRNA(‘transcriptomics’). • The actual ‘executives’ of the cell are the proteins, which perform myriad roles.
Introduction • Development of protein from mass spectrometry(MS) -based proteomics • The proteome of a cell designates the totality of all expressed proteins in a given biological situation (identity, amount, their state of modification, turnover, location, interaction partners, structures, functions) • in systems biology, it is usually the proteome that is the object of modeling
Introduction • The resulting proteomic data perfectly complement large-scale studies following individual proteins in single cells. • One of the most important areas for MS-based proteomics is the analysis of post-translational modifications(PTMs)
MS-based proteomics workflow • The field of MS-based proteomics has been made possible by seminal advances in technology that have helped to overcome a number of critical challenges ‘shotgun’ workflow
MS-based proteomics workflow Database search
MS-based proteomics workflow <outline of a typical shotgun proteomics workflow> A. Sample preparation
MS-based proteomics workflow B. Liquid chromatography – mass spectrometry
MS-based proteomics workflow C. Spectra interpretation
MS-based proteomics workflow • proteins or peptides could not be transferred into the vacuum of the mass spectrometer without being destroyed <Alternative approach> ESI(electrospray ionization) + MALDI(matrix-assisted laser desorption/ionization)
MS-based proteomics workflow ionization seperation detection MALDI(Matrix assistedlaserdesorption/ionization ) ESI(Electrospray ionization)
MS-based proteomics workflow • Biological sample used detergent-mediated solubilization(first MS sample preparation method) • Detergent-free protein(in-solution digestion method) • Stained protein band excised in-gel digested & analyzed in MS by MALDI or electrospray • Mass fingerprinting in conjuction with 2D-GE
MS-based proteomics workflow • proteomics experiments should be planned with the minimum number of fractionation steps possible. • Peptides spectrometer m/z & intensity • Key characteristics of a high performance mass spectrometer are resolution, mass, accuracy, speed, sensitivity and dynamic range
MS-based proteomics workflow • Shotgun approach(the use of bottom-up proteomics techniques in identifying proteins in complex mixtures) • Target approach(the mass spectrometer is fed with a list of predefined peptide species and their corresponding fragments) • Both shotgun and targeted approaches have their advantages and drawbacks
COMPUTATIONAL PROTEOMICS • An important aspect of high-throughput technologies is the availability of suitable computational workflows supporting the analysis and interpretation of the large-scale datasets • Of particular importance is the control of false positives • The computational steps that are needed to generate quantitative protein expression values from the raw data
COMPUTATIONAL PROTEOMICS <Overview of the main components of the computational workflow of shotgun proteomics> 1) feature detection and processing 2) peptide identification 3) Protein identification 4) quantification
COMPUTATIONAL PROTEOMICS Detection and processing of peptide features in LC-MS runs It is concerned with extracting features from the raw data that correspond to peptides in the MS spectra and to peptide fragments in the MS/MS spectra.
COMPUTATIONAL PROTEOMICS Identification of peptides based on their characteristic fragmentation patterns De novo approach Interpreting mass differences between fragment peaks as a.a Database search approach digests the protein sequences of an organism in-silico to obtain a list of peptides
COMPUTATIONAL PROTEOMICS Assembly of peptides into proteins It needs to be assigned to proteins, a non-trivial task that has been termed the ‘protein inference problem
COMPUTATIONAL PROTEOMICS the absolute or relative amounts of proteins in different samples usually need to be calculated, which requires quantification of the identified peptides.
INTERACTION PROTEOMICS • The dissection of molecular assemblies has been a longstanding goal of modern biology, which requires identification of the constituent partners as the first step. • All approaches in MS-based interaction proteomics are based on the assumption that a molecular interaction is the result of an affinity.(bait, prey)
INTERACTION PROTEOMICS AP-MS Generic scheme of the affinity purification-mass spectrometry workflow Quantitative comparison of the amounts of proteins in affinity purifications vs. control purifications distinguishes specific interactors from background binders Mass spectrometry is then used to identify the ‘prey’ proteins that interact with the bait.
INTERACTION PROTEOMICS • tandem affinity purification (TAP) : identification of the members of protein complex
INTERACTION PROTEOMICS Two-hybrid screening a molecular biology technique used to discover protein-protein interaction by testing for physical interactions
INTERACTION PROTEOMICS • the next challenge is to provide additional functionally relevant information such as topology and stoichiometry. The identification of cross-linked peptides yields spatial restraints that can be used to infer the topology of interactions and to map binding sites
INTERACTION PROTEOMICS Construction of interaction networks from large-scale AP-MS datasets. Ever-growing large-scale datasets will become increasingly useful for biologists and systems biologists alike
LARGE-SCALE DETERMINATION OF POST-TRANSLATIONAL MODIFICATIONS • Post-translational modifications (PTMs) of proteins are a key regulatory mechanism in signal transmission that controls nearly all aspects of cellular function. • cells also extensively use PTMs, which constitute an important class of molecular switches, for signal propagation to control the activity, structure, localization and interactions of proteins.
LARGE-SCALE DETERMINATION OF POST-TRANSLATIONAL MODIFICATIONS • PTMs should be identified and quantified in an unbiased and global manner. • PTM enrichment: Substoichiometric PTM-bearing proteins or peptides are enriched using various strategies
LARGE-SCALE DETERMINATION OF POST-TRANSLATIONAL MODIFICATIONS • PTM identification and localization: MS directly measures the presence of a PTM by a defined corresponding shift in mass of the peptide and PTM location within the peptide is obtained by the MS/MS pattern with single amino acid resolution.
LARGE-SCALE DETERMINATION OF POST-TRANSLATIONAL MODIFICATIONS • PTM site occupancy represents the fraction of a protein that is modified at a given PTM site. • Site occupancies can be calculated if one can quantify changing amounts of a modified peptide, the corresponding unmodified peptide and the entire protein in a perturbed system.
LARGE-SCALE DETERMINATION OF POST-TRANSLATIONAL MODIFICATIONS • large-scale PTM studies now serve as an information-rich resource to the community • biological researchers can focus on regulatory PTM sites • The data can also be used to investigate basic characteristics of particular PTMs,
OUTLOOK AND FUTURE CHALLENGES • MS-based proteomics advances affect the entire proteomics workflow, starting with sample preparation and ending with computational proteomics • The advent of high-resolution high-accuracy MS data, combined with sophisticated quantification strategies, has been especially important in obtaining biologically relevant information from MS-based proteomics
OUTLOOK AND FUTURE CHALLENGES Unique contribution of different ‘omics’ technologies to systems biology. A. The sequences of genomes and their epigenetic marks for many genomes in parallel. B. The gene expression program and the comparison of changes of gene expression C. A complete picture of all proteins, the primary agents of cellular processes.
OUTLOOK AND FUTURE CHALLENGES • Proteomics can give us a detailed picture of the end product of the gene expression cascade, the mature, active and fully modified protein form. • it can characterize gene expression at subcellular resolution. • MS-based proteomics is crucial to a systems-level understanding of cellular function.