Download
1 / 39
390 likes | 546 Views
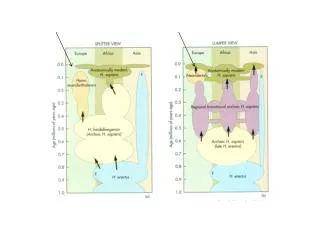
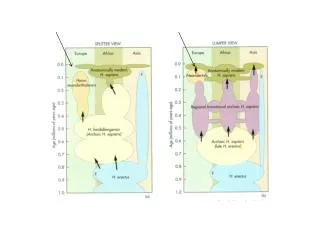
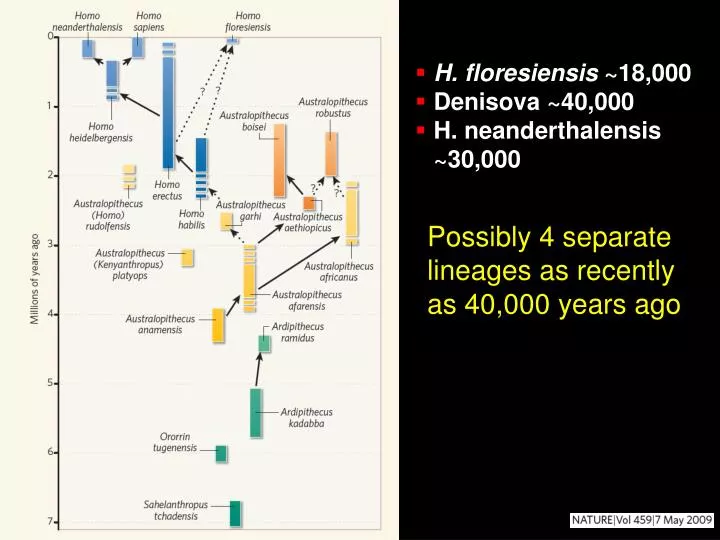
H. floresiensis ~18,000 Denisova ~40,000 H. neanderthalensis ~30,000. Possibly 4 separate lineages as recently as 40,000 years ago. RECONSTRUCTING THE PATH OF HUMAN MIGRATION. Cave Paintings ~35,000 years ago. 35,000 year-old statues. More….

E N D
H. floresiensis ~18,000 • Denisova ~40,000 • H. neanderthalensis ~30,000 Possibly 4 separate lineages as recently as 40,000 years ago
RAPID INCREASE IN TECHNOLOGY, ART, AND MUSIC DEMONSTRATES CULTURAL EVOLUTION IN MODERN HUMANS.
HUMANS ARE UNIQUE IN THEIR EXTREMELY HIGH RATE OF CULTURAL EVOLUTION • To what extent is an interaction between cultural evolution and phenotypic evolution possible?
Early arrivals. New evidence, including stone tools apparently used for clearing trees (inset), shows humans were in the Ivane Valley from 43,000 to 49,000 years ago and later migrated to offshore islands.
D-loop region Human mtDNA
The patterns of mtDNA variation have been useful in reconstructing the historical pattern of migration in the Malayo-Polynesian and Oceanic islands.
Invasion of North America Clovis sites
Single OR Multiple Invasions of the Americas • Only opened about 13,000 years ago
The Late Pleistocene Dispersal of Modern Humans in the Americas Goebel et al. Science 14 March 2008: Vol. 319. no. 5869, pp. 1497 - 1502
ALTERNATE HYPOTHESES FOR THE PEOPLING OF THE AMERICAS From: Dillehay Nature 2003
Variation in the Human D-loop Region of the mtDNA Haplotype Groups
Oldest American genetic sample reveals early New World frontiers. The oldest sample of human DNA ever isolated in the Americas is providing a glimpse of how people spread across the land masses. The DNA was extracted from teeth, more than 10,000years old, found in a cave on the northern tip of Prince of Wales Island, off southern Alaska. Researchers compared the pattern of mutations in the DNA against those in thousands of samples. They found matches with 47 Native Americans from tribes living in areas ranging from North America to Tierra del Fuego, showing how the caveman's descendants must have spread. The caveman belonged to 'lineage D', one of the five founding lineages believed to have settled in the Americas more than 10,000 years ago. Lineage D is thought to have originated in Asia, and researchers also found a close match with a member of the Han ethnic group from Qingdao in eastern China. Nature436, 162 (14 July 2005)
] “Amerindian” • Worldwide Human Relationships Inferred from Genome-Wide Patterns of Variation • 938 individuals • 51 populations • 650,000 SNPs ] African Li et al. Science 22 February 2008:Vol. 319. no. 5866, pp. 1100 - 1104
Polymorphism and Variation in Human Genomes: Single Nucleotide Variants (SNPs) Current estimates are that SNPs occur as frequently as every 100-300 bases. This implies in an entire human genome there are approximately 10 to 30 million potential SNPs. More than 4 million SNPs have been identified and the information has been made publicly available through the efforts of TSC and others. Many of these SNPs have unknown associations. Compilation of public SNPs by NCBI has produced a subset of SNPs defined as a non-redundant set of markers that are used for annotation of reference genome sequence and are thus referred to as reference SNPs (rsSNPs). Over 2.6 million SNPs have currently been assigned as "rsSNPs". Recent work has suggested that about 10 million SNPs that are common in human population are not inherited independently; rather, sets of adjacent SNPs are present on alleles in a block pattern, so called haplotype. Many haplotype blocks in human have been transmitted through many generations without recombination. This means although a block may contain many SNPs, it takes a few SNPs to identify or tag each haplotype in the block.
Advancing RNA-Seq analysis Haas & Zody Nature Biotechnology (2010) 28: 421–423
SNPs and Diagnostics Eventually, SNP profiles that are characteristic of a variety of diseases will be established. Then, it will only be a matter of time before physicians can screen individuals for susceptibility to a disease just by analyzing their DNA samples for specific SNP patterns. Many common diseases in humans are not caused by one genetic variation within a single gene, but are determined by complex interactions among multiple genes, environmental and lifestyle factors. Genetic factors confer susceptibility or resistance to a disease and influence the severity or progression of disease. Since we do not yet know all of the factors involved in these intricate pathways, researchers have found it difficult to develop screening tests for most diseases and disorders, such as diabetes, many cardiovascular diseases, Alzheimer's disease, arthritis, to name just a few. By studying SNP profiles or haplotypes associated with a disease trait, researchers may begin to reveal relevant genes associated with a disease. Association study can detect and indicate which pattern is most likely associated with the disease-causing genes. Eventually, SNP profiles that are characteristic of a variety of diseases will be established. Then, it will only be a matter of time before physicians can screen individuals for susceptibility to a disease just by analyzing their DNA samples for specific SNP patterns.
Two challenges for human disease research: • Linking these vast data to genes and functional elements in the human genome • Linking polymorphisms in the human genome to disease phenotypes
The new Omni5 harnesses over ten years of genomic research with more than 4.3 million high-value markers. And room for 500k of your own.
Discovery of variation and association with Disease phenotypes • Linking variation to function
Medical Genomics “Nothing in biology makes sense except in light of evolution” Theodosius Dobzhansky 1973