Download
1 / 1
10 likes | 142 Views
Quantum Chemical Studies of Energetics of RNA-Drug Interactions. Anubhooti , Purshotam Sharma, Gopalakrishnan Bulusu and Abhijit Mitra Center for Computational Natural Sciences and Bioinformatics (CCNSB), International Institute of Information Technology, Hyderabad-500032. Introduction.

E N D
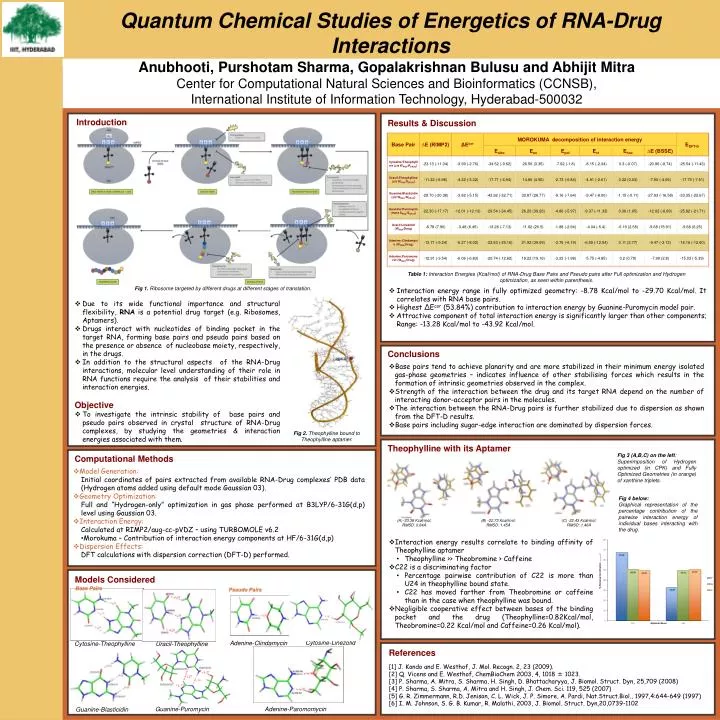
Quantum Chemical Studies of Energetics of RNA-Drug Interactions Anubhooti, Purshotam Sharma, GopalakrishnanBulusuand AbhijitMitra Center for Computational Natural Sciences and Bioinformatics (CCNSB), International Institute of Information Technology, Hyderabad-500032 Introduction Results & Discussion Table 1: Interaction Energies (Kcal/mol) of RNA-Drug Base Pairs and Pseudo pairs after Full optimization and Hydrogen optimization, as seen within parenthesis. Fig 1. Ribosome targeted by different drugs at different stages of translation. • Interaction energy range in fully optimized geometry: -8.78 Kcal/mol to -29.70 Kcal/mol. It correlates with RNA base pairs. • Highest ΔEcor (53.84%) contribution to interaction energy by Guanine-Puromycin model pair. • Attractive component of total interaction energy is significantly larger than other components; Range: -13.28 Kcal/mol to -43.92 Kcal/mol. • Due to its wide functional importance and structural flexibility, RNA is a potential drug target (e.g. Ribosomes, Aptamers). • Drugs interact with nucleotides of binding pocket in the target RNA, forming base pairs and pseudo pairs based on the presence or absence of nucleobase moiety, respectively, in the drugs. • In addition to the structural aspects of the RNA-Drug interactions, molecular level understanding of their role in RNA functions require the analysis of their stabilities and interaction energies. • Objective • To investigate the intrinsic stability of base pairs and pseudo pairs observed in crystal structure of RNA-Drug complexes, by studying the geometries & interaction energies associated with them. Conclusions • Base pairs tend to achieve planarity and are more stabilized in their minimum energy isolated gas-phase geometries – indicates influence of other stabilising forces which results in the formation of intrinsic geometries observed in the complex. • Strength of the interaction between the drug and its target RNA depend on the number of interacting donor-acceptor pairs in the molecules. • The interaction between the RNA-Drug pairs is further stabilized due to dispersion as shown from the DFT-D results. • Base pairs including sugar-edge interaction are dominated by dispersion forces. Fig 2. Theophylline bound to Theophylline aptamer. Theophylline with its Aptamer • Model Generation: • Initial coordinates of pairs extracted from available RNA-Drug complexes’ PDB data (Hydrogen atoms added using default mode Gaussian 03). • Geometry Optimization: • Full and “Hydrogen-only” optimization in gas phase performed at B3LYP/6-31G(d,p) level using Gaussian 03. • Interaction Energy: • Calculated at RIMP2/aug-cc-pVDZ – using TURBOMOLE v6.2 • Morokuma – Contribution of interaction energy components at HF/6-31G(d,p) • Dispersion Effects: • DFT calculations with dispersion correction (DFT-D) performed. Fig 3 (A,B,C) on the left: Superimposition of Hydrogen optimized (in CPK) and Fully Optimized Geometries (in orange) of xanthine triplets. Computational Methods Fig 4 below: Graphical representation of the percentage contribution of the pairwise interaction energy of individual bases interacting with the drug. • -33.36 Kcal/mol; • RMSD: 0.84Å (B) -22.73 Kcal/mol; RMSD: 1.45Å (C) -22.43 Kcal/mol; RMSD: 1.46Å • Interaction energy results correlate to binding affinity of Theophylline aptamer • Theophylline >> Theobromine > Caffeine • C22 is a discriminating factor • Percentage pairwise contribution of C22 is more than U24 in theophylline bound state. • C22 has moved farther from Theobromine or caffeine than in the case when theophylline was bound. • Negligible cooperative effect between bases of the binding pocket and the drug (Theophylline=0.82Kcal/mol, Theobromine=0.22 Kcal/mol and Caffeine=0.26 Kcal/mol). Models Considered Base Pairs Pseudo Pairs Cytosine-Linezolid Adenine-Clindamycin Cytosine-Theophylline Uracil-Theophylline References [1] J. Kondo and E. Westhof, J. Mol. Recogn. 2, 23 (2009). [2] Q. Vicens and E. Westhof, ChemBioChem 2003, 4, 1018 ± 1023. [3] P. Sharma, A. Mitra, S. Sharma, H. Singh, D. Bhattacharyya, J. Biomol. Struct. Dyn, 25,709 (2008) [4] P. Sharma, S. Sharma, A. Mitra and H. Singh, J. Chem. Sci. 119, 525 (2007) [5] G. R. Zimmermann, R.D. Jenison, C. L. Wick, J. P. Simore, A. Pardi, Nat.Struct.Biol., 1997,4:644-649 (1997) [6] I. M. Johnson, S. G. B. Kumar, R. Malathi, 2003, J. Biomol. Struct. Dyn,20,0739-1102 Guanine-Puromycin Adenine-Paromomycin Guanine-Blasticidin