Download

1 / 18
190 likes | 395 Views
Spinocerebellar Ataxia and X-Linked Bulbospinal Neuronopathy. Trinucleotide repeat expansions and other dynamic mutations Roy Poh October 2007. Spinocerebellar Ataxias. Autosomal dominant neurodegenerative disorder Genetically heterogeneous 28 subtypes listed

E N D
Spinocerebellar Ataxiaand X-Linked Bulbospinal Neuronopathy Trinucleotide repeat expansions and other dynamic mutations Roy Poh October 2007
Spinocerebellar Ataxias • Autosomal dominant neurodegenerative disorder • Genetically heterogeneous • 28 subtypes listed • 9 SCAs are caused by dynamic mutations - SCA1, 2, 3, 6, 7 & 17 - due CAG repeat expansion also referred as polyglutamine disorder. - SCA8 (CTG repeat), SCA10 (ATTCT repeat), SCA12
Clinical features of SCAs • Characterised by progressive cerebellar ataxia of gait and limbs • With variably associated with extrapyramidal & pyramidal signs, eye movements, deafness, retinopathy, peripheral neuropathy • Disease onset is usually between 30 and 50 (Childhood and adult >60 years old has been reported) • Difficult to classify based on clinical features only
Classification of SCAs • Use as guideline for clinical practise and prioritize genetic tests for diagnosis.
Prevalence • Up to 5-7 in 100,000 in some populations • Frequency varies in population and ethically • Founder effects contribute to variable prevalence in subtypes Worldwide SCA 3 21% SCA 2 15% SCA 6 15% SCA 7 5% SCA 1 6% SCA 8 3% Rare 5% Unknown 30%
Genetics of SCA • Autosomal dominant • CAG repeat expansion vary among disorders - A strong correlation between number of repeats and severity of the disease (larger number of repeats – earlier onset and more severe of the disease) - Mutable normal alleles (intermediate alleles) (Increased risk of disease causing allele in next generation; reduced / full penetrance?) • Anticipation – earlier onset and increase severity in next generation; somatic instability (in larger repeats), SCA7 being most unstable • Repeat expansion outside coding region (5’UTR, Intron, 3’UTR) • Missense mutations
Genetic tests • Estimated 50 - 60% can be identified (SCA1, 2, 3, 6, 7, 8, 10, 12 , 17 and DRPLA) • Test available not associated with repeat expansions (SCA 5, 13, 14, 27, 16q22-linked SCA) • Test in group – based on broad clinical overlap - eg. Common group - SCA 1, 2, 3, 6 & 7 Follow by - SCA 10, 12, 14 & 17 • PCR Analysis • Southern Blot analysis - SCA 2, 7, 8 & 10 (> 100 CAG repeats) • Individualized testing (eg. specific diagnosis, ethic background and population frequency)
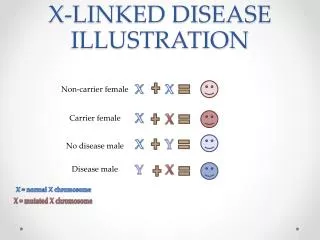
X-Linked Bulbospinal neuronopathy • Kennedy disease / Spinal & Bulbar Muscular Atrophy (SBMA) • Polyglutamine disorder (CAG in exon 1 of Androgen receptor on Xq11-12) • X-linked recessive and affect only males • Few than1:50,000 live male births (Seen Caucasian or Asian but not in African or Aboriginal racial background) • Symptoms begin age 20 to 50 years and not in childhood and adolescence • Mutation in AR gene cause androgen insensitivity syndrome
Clinical Features • Slow progressive neuromuscular disorder • Degeneration of lower motor neuron results in proximal muscle weakness, muscular atrophy and fasiculations. • Affected individuals show gynecomastia, testicular atrophy and reduced fertility as a result of mild androgen insensitivity • Female carriers with expanded CAG usually asymptomatic but muscle cramps and occasional tremors reported
Clinical features (cont) • Often confused with: - Amyotrophic lateral sclerosis (ALS) - 1 in 25 cases - Spinal muscular atrophy type 4 (SMA) - SCA 3 - Friedreich ataxia
Genetic test • PCR amplification of CAG repeat in AR gene • Normal allele: ≤34 CAG repeats • Full penetrance allele: ≥38 CAG repeats • Reduced penetrance allele: 36 - 37 CAG repeats (35 CAG repeats – ?, no consensus) • CAG repeat relatively stable with small length shifts and contractions • Generally repeat length inversely correlates with ages of onset but exceptions have been reported • Genotype/phenotype correlation in 60% of cases
Polyglutamine disorders – Mechanisms • Molecular and cellular events that underlie neurodegeneration is still poorly understood • Mechanisms proposed: - Polyglutamine tract is proteolytically cleavaged releasing toxic fragments and inclusions formation – (except SCA 2 & 6 where found in cytoplasm) - Expanded polyglutamine tract leads to conformational change and aggregation - Expanded polyglutamine tract causes sequestration of cellular factors
Mechanisms (con’t) • Findings indicate Interference with molecular pathways: - Protein aggregation and clearance - Ubiquitin-proteasome system - Transcriptional regulation - Alterations of calcium homeostasis • Pathways could act independently or interact and enhance each others • Leading to neuronal dysfunction and death • Disease causing genes are expressed widely in affected neurons only
Summary: Polyglutamine disorders • SCA1, 2, 3, 6, 7, 17 and SBMA (DRPLA & HD) • CAG repeat in the coding region of the gene • Overlapping Clinical pathological features • SBMA is relatively distinct from SCAs • Encoded proteins are unrelated and without similarity in functions and sub-cellular localisation • Pathogensis linked to polyglutamine expanded tract – causes toxic gain of function but mechanism not completely known
Reference • www.geneclinics.org • Autosomal dominant cerebellar ataxia: clinical features, genetics and pathogenesis. Lancet Neurology, 2004, 3, 291-304. • Molecular Pathogeneisis of spinocerebellar ataxia. Brain, 2006, 129, 1357-1370. • Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Human Molecualr Genetics, 2007, 16, 115-123. • Repeats instability: Mechanisms of dynamic mutations. Nature Review Genetics, 2005, 6, 729-742. • Therapeutics development for triplet repeat expansion diseases. 2005, Nature Review Genetics, 6, 756-765.