Download

1 / 30
310 likes | 702 Views
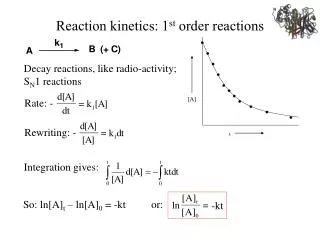
•. •. •. •. •. •. •. •. [A]. •. •. •. t. Reaction kinetics: 1 st order reactions. Decay reactions, like radio-activity; S N 1 reactions. Rate: -. Rewriting: -. Integration gives: . So: ln[A] t – ln[A] 0 = -kt or: . -kt.

E N D
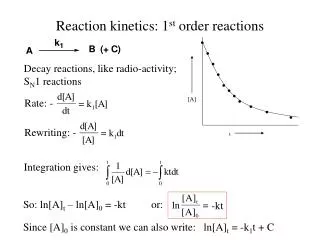
• • • • • • • • [A] • • • t Reaction kinetics: 1st order reactions Decay reactions, like radio-activity; SN1 reactions Rate: - Rewriting: - Integration gives: So: ln[A]t – ln[A]0 = -kt or: -kt Since [A]0 is constant we can also write: ln[A]t = -k1t + C
Plotting ln[A]t against t gives a straight line with slope -k: -kt The halflife, t1/2, is defined as the time that is needed to reduce the concentration of the reactant to 50% of its original value. In formula: =-kt1/2 so
Reaction kinetics: 2nd order reactions So: - = - When [A] [B], this equation is mathematically rather complicated.A simplification reads as follows: take [P] = x, then [A] = [A]0 – x and [B] = [B]0 – x The rate then becomes: n = k2([A]0-x)([B]0-x) so Integration gives: Plotting against t gives a straight line with slope k2([B]0-[A]0)
Special cases: • [A]0>>[B]0 (pseudo-first order kinetics) Example: - in which k'=k2[H2O] This is a pseudo-first order reaction, since [H2O] is constant. The second-order rate constant k2 can be calculated from k' and [H2O]. In a dilute aqueous solution, [H2O]=55 M.
Special cases: • [A]0 = [B]0 Integration gives: Plotting of against t gives k2 as the slope.
[A]0 [A]t xe [B]t t Reversible reactions Take the simplest possibility: On t = 0: [A] = [A]0 [B] = 0 t = t: [A] = [A]0-x [B] = x = k1[A] – k-1[B] = k1([A]0 – x) – k-1x = k1[A]0 – (k1 + k-1)x Integration gives: (1) At equilibrium, the net reaction rate = 0, so [B]t is constant (=[B]e = xe), so: k1[A]e = k-1[B]e = k-1xe
There is an equilibrium constant: so: (2) Combining eq (1) with (2) gives: This is the rate equation for a first order process! Determination of (k1 + k-1) by plotting against t Eq (2) gives 2 equations, 2 unknowns Individual values of k1 and k-1 can be determined
Preequilibria Very complicated kinetics, unless you assume that [A·B] is constant during a large part of the reaction (steady state approach) k1[A][B] = k-1[A·B] + k2[A·B] = (k-1 + k2)[A·B] So the rate equation now becomes: n = k2[A·B] =
A0 A0 [C] [A·B] [C] [A·B] xe t n = k2[A·B] = Two possibilities: - rapid breakdown of A·B, k2>>k-1, so n = k1[A][B] t - slow breakdown of the complex: k2<<k1,k-1, so: n = k2[A·B] = = k2K[A][B]
X‡ (TS) Eact E S P Reaction coordinate Interpretation of rate constants:the Arrhenius equation Every reaction has to overcome an energy barrier: the transition state (TS, X‡). At higher temperature, more particles are able to overcome the energy barrier. Arrhenius equation: Ea can be determined by measuring kobs at two different temperatures:
Idem, from statistical mechanics (collision theory) Arrhenius: P = probability factor (not every collision is effective) Z = collision number (number of collisions per second)
X‡ (TS) Eact E S P Reaction coordinate Idem, from transition state theory: or [X‡] = K‡[A][B] n = k‡[X‡] = k‡K‡[A][B] = k[A][B], so k = k‡K‡ Statistical mechanics gives us the following relation: kB = Boltzmann’s constant; h = Planck’s constant so
For all equilibria we can write: DG0 = - RT ln K, so for our case we get: DG‡ = - RT ln K‡ Expressing K‡ in terms of DG‡ and RT gives the following equation for k: (1) Since DG‡ = DH‡ - TDS‡, we can also write: (2) Eq (1) and (2) are called the Eyring equations
ln A slope = ln k 1/T slope = 1/T The Eyring and Arrhenius equations resemble each other: Arrhenius: so: so Ea = DH‡ + RT Eyring: In order to determine DH‡ and DS‡ it is easier to differentiate ln (k/T) to 1/T:
So, the procedure to determine activation parameters is: - determine k at different temperatures - plotting ln(k/T) against 1/T gives DH‡ - then gives DS‡ and when you have DH‡ and DS‡, you also have DG‡ since DG = DH-TDS
Interpretation of activation parameters • DG‡, the Gibbs free energy of activation, determines at which rate a certain reaction will run at a given temperature • DH‡ is a measure for the amount of binding energy that is lost in the transition state relative to the ground state (including solvent effects) • DS‡ is a measure for the difference in (dis)order between the transition state and the ground state • for monomolecular reactions: DS‡ 0 J/mol.K • for a bimolecular reaction: DS‡ << 0 J/mol.K(two particles have to come together in the transition state to form one particle, demanding a much greater order)
Example: DG‡ = 62.8 kJ/mol (very fast rx) DH‡ = 33.0 kJ/mol (rel. low, compensation of C-H bond cleavage by hydration TS) DS‡ = -100 J/mol.K (bimolecular rx)
Another example: DH‡ = 85 kJ/mol (relatively high: no new bonds to be formed, no compensation for the partial cleavage of the C-C bond in the transition state; acetonitrile is aprotic, compensation of DH‡ by solvation will be less than in water DS‡ = 0 J/mol.K (monomolecular reaction)
Application of activation parameters for the elucidation of reaction mechanisms: A DS‡ of +12 J/mol.K was found monomolecular process A DS‡ of -117 J/mol.K was found bimolecular process; rate determining step in this case is the attack of water on the carbonyl group. Look in your course book for the exact reaction mechanisms!
Solvation (solvent effects) Influence of solvation on the reaction rate: k(H2O) = 10-7 l.mol-1.s-1; k(DMF) = 10-1 l.mol-1.s-1 so DDG‡ ~ 30 kJ/mol DG‡DMF DG‡H2O DMF E H2O DG‡DMF < DG‡H2O reaction progress
What is the background of this strong solvent effect? In H2O there is more solvation than in DMF, due to hydrogen bonds. Note the changes in entropy: loss of DS‡ because of orientation of the substrates, gain of DS‡ because of the liberation of water (less solvated transition state). The balance is not easy to predict! In general, in case of ions, the ground state is more solvated than the transition state: TS (‡) is hardly solvated due to the spreading of charge. Again a strong solvent effect here: k(H2O)= 7.4x10-6 s-1; k(DMF) = 37 s-1
Solvation effects in (bio)polymers Polymers or enzymes may have apolar pockets, which leads to: - less solvation and therefore higher reaction rates; - changes in pKa’s of acidic/basic groups: [PyN][H+] R = CH3: pKa = 9.7 R = polymer: pKa = 7.7 Ka = [PyNH+] E.g. lysine, R-NH2 + H+ R-NH3+ pKa (H2O) = 10.4, in some enzymes pKa = 7 !
The energy diagram Consider the gas phase chlorination of methane: CH4 + Cl• rH-Cl H3C···H···Cl Reaction course is via the route of lowest energy (“mountain pass”) CH3• + HCl rC-H
A cross-section of this “mountain landscape” gives the well-known energy diagram: H3C···H···Cl
What does the transition state look like? Hammond postulate: The TS closely resembles the species with the highest energy content Exothermic reaction (a) has a low Ea, TS resembles ground state; Endothermic reaction (b) has a high Ea, TS resembles the product
Kinetic isotope effects Difference in effectivity of C-H and C-D bond cleavage: primary isotope effect The background is the difference in bond strength, caused by the difference in mass between H and D: E0 of covalent bond is given by: 1/2hn = 1/2hc(1/l); 1/l(C-H) 3000 cm-1, E0(C-H) 18 kJ/mol; 1/l(C-D) 2200 cm-1, E0(C-D) 13 kJ/mol
In this case: Ea = D kobs = x 7.5 This is the maximum primary kinetic isotope effect at ~25ºC. The isotope effect tells us something about the transition state. For this, we have to look at the stretching vibrations of the C-H(D) bond: Antisymmetrical stretching vibration: leads to reaction Symmetrical stretching vibration: involvement of H(D) depends on the structure of the transition state
a b c EaH E0‡ E0‡ E0C-H E0‡ E0C-D EaH EaD EaH EaD EaD E E0C-H E0C-H E0H-Cl E0C-D E0D-Cl E0C-D reactants products H3C · · · · ·H · · · · ·Cl H3C · · H · ···· · · ·Cl H3C · · · · · · · · H · ·Cl Symmetrical stretching vibration: involvement of H(D) depends on the structure of the transition state: H exactly in the middle between C and Cl: symmetrical transition state, kinetic isotope effect is maximum (~7.5). H not in the middle: isotope effect < 7.5 late TS symmetrical TS early TS
Rule of thumb: kH/kD 4-7: bond cleavage, symmetrical transition state kH/kD 1-4: bond cleavage, asymmetrical transition state; or no bond cleavage (secondary isotope effect) Example: Maximum isotope effect at symmetrical TS, so when pKa(acid) = pKa(HB)