Download
1 / 23
740 likes | 3.59k Views
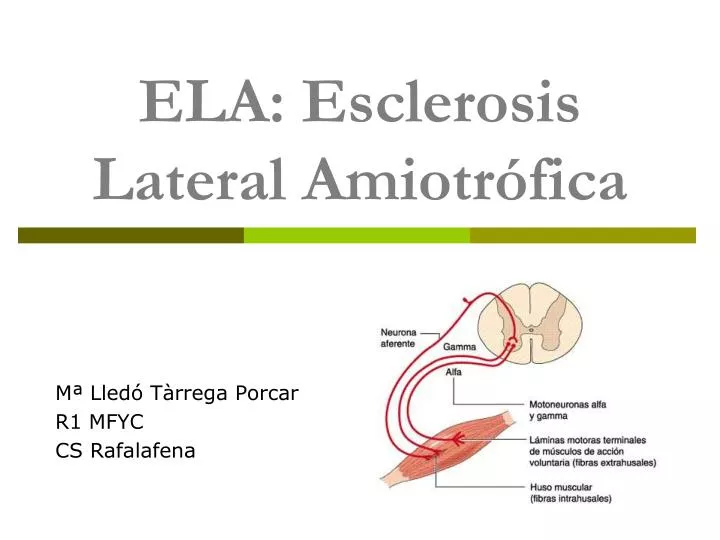
ELA: Esclerosis Lateral Amiotrófica. Mª Lledó Tàrrega Porcar R1 MFYC CS Rafalafena. INTRODUCCIÓN . Descrita por primera vez en 1869 por el médico francés Jean Martin Charcot (1825-1893). También llamada enfermedad de Lou Gehrig y, en Francia, enfermedad de Charcot .

E N D
ELA: Esclerosis Lateral Amiotrófica Mª Lledó Tàrrega Porcar R1 MFYC CS Rafalafena
INTRODUCCIÓN • Descrita por primera vez en 1869 por el médico francés Jean Martin Charcot (1825-1893). • También llamada enfermedad de Lou Gehrig y, en Francia, enfermedad de Charcot. • Definición: Enfermedad degenerativa de las motoneuronas superiores e inferiores de tipo neuromuscular, que provoca una parálisis muscular progresiva y en las fases finales tiene un pronóstico mortal.
Clasificación de las enfermedades de las motoneuronas: • Enfermedades de las motoneuronas inferiores: Atrofias espinales:a) Poliomielitis aguda, sd. Postpolio.b) Neuropatías motoras hereditarias. c) Atrofia bulboespinal (enfermedad de Kenedy) ligada al cromosoma X.d) Atrofia muscular espinal infantil, juvenil y del adulto (AME). • 2) Enfermedades de las motoneuronas superiores (corticales):a) Esclerosis lateral primaria. b) Paraplejía espástica hereditaria.c) Paraparesia espástica tropical (HTLV-1). d) Neurotóxicas (latirismo y konzo). Otras. • 3) Esclerosis lateral amiotrófica (motoneuronas superiores e inferiores):a) Esporádica: variedad clásica (asociada a la degeneración de otros sistemas y/o demencia). b) Familiar (herencia variable): mutaciones en el gen SOD-1, cromosoma 17, otras. c) Variedad de la isla de Guam.
1. Epidemiología: · Prevalencia:4.6/100.000 habitantes. · Incidencia:0.4 – 1.8/100.000 hab./año. - Relación H/M: 3/1 • Afectación predominante de los 40 a los 70 años. Raramente se producen casos antes de los 30 años de edad y después de los 70. • Supervivencia media: 3 años. (Ésta depende sobretodo de la precocidad con la que se afecte la respiración. También influye la edad de comienzo, siendo más mortal en individuos más añosos). < 45 años: supervivencia de 54.8 meses. > 45 años: supervivencia de 25.4 meses. • ELA bulbar: < 1 año de supervivencia.
2. Etiopatogenia: • Etiología:desconocida (90%), casos esporádicos, sin demostrarse claramente etiología hereditaria. 10% de los casos tienen origen genético. • Factores desencadenantes: Se han buscado agente ambientales, neurotóxicos y otros, sin llegar a ninguna conclusión, incluso en estudios poblacionales (península de kii, isla de Guam). • ELA esporádica de Occidente: genes de susceptibilidad de la Hemocromatosis y SMN 1. • Factores locales bioquímicos: estrés oxidativo, daño excitotóxico por glutamato, alteración de la homeostasis de los productos del citoesqueleto, mala regulación del calcio intracelular, disfunción del metabolismo mitocondrial, etc. Se ha postulado que estos factores podrian ser posibles DIANAS TERAPÉUTICAS para evitar la neurodegeneración.
3. Anatomia Patológica (I): · Degeneración de las motoneuronas superiores: • No todos los grupos neuronales medulares son igual de sensibles a la degeneración, distinguimos: - Resistentes: El núcleo de Onuf en los segmentos sacros, es muy resistente, esto explica la conservación tardía de la inervación esfinteriana. También son muy resistentes el facial, trigémino y oculmotores. - Sensibles:núcleo ambiguo e hipogloso: de ahí la frecuencia e intensidad de la parálisis bulbar. • Neuronas superiores, signos de atrofia: citoplasmas retraidos y núcleos pignóticos. (Algunas neuronas presentan aspecto de degeneración walleriana).En los núcleos motores afectados y el asta anterior de la médula, se observa una evidente pérdida neuronal y reacción glial.
Anatomía Patológica (II): • Pequeñas inclusiones eosinófilas positivas para ubiocuitina en núcleos motores medulares y de la corteza frontal e hipocampo. • Algunas variedades de ELA se asocian a demencia frontotemporal, dependiendo de la positividad a diferentes tipos de inclusiones neuronales (ubicuitina, Tau, a-sinucleina) y asociaciones entre ellas. · Degeneración de las motoneuronas inferiores: • Atrofia macroscópica de las raíces anteriores medulares. • Músculos: se observa un patrón de atrofia neurógena con fibras angulosas agrupadas.
4. Genética: 5-15% casos de ELA familiar, con base genética heterogénea, en la cual se han descrito ligamientos a varios loci, pero solo se han descrito 4 genes: • 1. ALS 1 o SOD-1 (gen de la superóxido dismutasa Zn/Cu): mutación más frecuente, 20% de ELA familiares. Relacionado con los mecanismos de protección de las células frente a los radicales libres. • 2. ALS 2 (proteina alsina): implicada en la supervivencia neuronal a través de una interacción con receptores de glutamato. También implicado en otras enfemedades neurológicas como Sd. De motoneurona superior, Paraplejía espática hereditaria o variedad infanto-juvenil de ELA). • 3. ALS 4/SETX: mutaciones en este gen dan lugar a una variedad muy rara de comienzo juvenil y rara evolución. • 4. ALS 8/VAPB (synaptobrevin-associated membrane protein B): primeras mutaciones descritas en varias grandes familias brasileñas con un ancestro común con herencia AD.
5. Clínica (I): Se distinguen 3 formas de comienzo de ELA: • Atrofia muscular progresiva (por lesión motora espinal). • Parálisis bulbar progresiva: de comienzo con disartria y disfagia. • Combinación de lesión de motoneuronas superior e inferior desde el inicio. En cuanto a la evolución y a la anatomía patológica, las 3 formas casi siempre se acaban confundiendo.
Clínica (II): Presencia de signos y síntomas de las 2 motoneuronas: • Signos y síntomas Motoneurona Superior: • Pérdida destreza (lentitud al ejecutar movimientos) • Pérdida fuerza muscular (debilidad) • Espasticidad (tensión muscular al estiramiento) • Hiperreflexia y reflejos patológicos (Babinski) • Parálisis pseudobulbar (risa y llanto espontáneo e inmotivado, afectación bulbar con disfagia, disartria) • Espasmos flexores (movimientos bruscos piernas) • Signos y síntomas Motoneurona Inferior: • Pérdida de fuerza muscular (debilidad) • Pérdida de masa muscular (atrofia) • Hipotonía • Hipo o arreflexia • Fasciculaciones: contracciones muscules espontáneas. • Calambres: mov. abruptos, involuntarios y dolorosos del músculo.
Clínica (III): • Síntomas iniciales: calambres, fasciculaciones, espasmos, sacudidas o debilidad. Fasciculaciones de la lengua o el mentón producen disatria o disfagia y son un signo de afectación bulbar. • Otra forma de comienzo: parálisis laríngea (disfonía espástica y estridor), con debilidad respiratoria (ELA como diagnóstico diferencial de la hipoventilación alveolar). • Otros síntomas/signos: con el tiempo se produce una anormal pérdida de masa muscular o de peso corporal (por dificultad para la alimentación), lo cual asocia una evolución desfavorable. • Progresión: irregular, asimétrica, progresa de forma distina en cada parte del cuerpo.Suele tener progresión centrípeta a toda la extremidad, y después la contralateral. Después se afectan el resto de los miembros.
Clínica (IV): • Variedad clásica: casi siempre comienza por una mano, que suele ser la dominante, con amiotrofia y pérdida de fuerza. • IMPORTANTE: una mano con amiotrofia global sin dolor ni trastornos sensitivos asociados anuncia con gran probabilidad una enfermedad de la motoneurona generalizada. • Otras variantes: comienzan con debilidad en los músculos de las cinturas y simulan una distrofia muscular o poliomielitis. También puede comenzar por las piernas y simular una polineuropatía o lesión de la cola de caballo.
Clínica (V): · Funciones respetadas: • Funciones cerebrales no relacionadas con la actividad motora: la SENSIBILIDAD y la INTELIGENCIA se mantienen inalteradas. • Motoneuronas que controlan los músculos extrínsecos del ojo: se conservan los movimientos oculares hasta el final. • Núcleo de Onuf: se conservan los músculos de los esfínteres que controlan la micción y la defecación.
6. Diagnóstico (I): • Hª clínica y exploración. Curso de la enfermedad. • Laboratorio: hemograma, bioquímica que incluya CK, VSG, RPR, hormonas tiroideas, IEF, B12. • Rx tórax • Si solo afectación MNI: Ac antiGM1. • Electrofisiología: a)confirmar alteración MNI en las regiones clínica/ afectadas, b) confirmar extensión a las clínica/ no afectadas, c) excluir otros procesos patológicos) d) descartar bloqueos o desmielinización. • RM encefálica y espinal: descartar otras causas.
Diagnóstico (II): • El diagnóstico de ELA requiere: • A.1) Evidencia de afectación MN inferior (MNI), clínica o electrofisiológica. • A.2) Evidencia de afectación de MN superior en el examen clínico. • A.3) diseminación progresiva de los síntomas, de una región a otra, determinada por la Hª o el examen. • Junto con la ausencia de: • B.1) Evidencia electrofisiológica o patológica de otra enfermedad que pudiera explicar los signos de degenerac. de MNI o MNS, y • B:2) evidencia de otra enfermedad en la neuroimagen que pudiera explicar los signos clínicos y electrofisiológicos observados. EMG típica: denervación activa en los músculos clínicamente afectados + conservación de las velocidades de conducción motora y sentiviva.
Diagnóstico (III): A favor del diagnóstico de ELA se encuentran: • La ausencia de dolory de alteraciones sensitivas. • La función normal de los esfínteres anal y vesical. • Los resultados normales de los estudios radiográficos de la columna y la ausencia de alteraciones en el líquido cefalorraquídeo. Imprescindible descartar todos los casos potencialmente tratables. Esto es importante sobre todo en casos atípicos por presentar: • Limitación de la enfermedad a la neurona motora superior o a la inferior. • Afección de neuronas distintas de las neuronas motoras. • Signos de bloqueo de la conducción nerviosa motora en el estudio electrofisiológico.
7. Diagnóstico diferencial: • Tumores (RM) • Siringomielia, siringobulbia (RM) • Mielopatía cervical espondiloartrósica (RM) • Neuropatía motora multifocal (EMG) • En casos bulbares miastenia gravis • Todos los Dx dif de ELP • Todos los DX de AMP Procesos que pueden imitar una ELA parcial o totalmente reversibles: • Intoxicación por Mercurio y Plomo • Mielopatía por carencia de cobre • Linfoma • Paraproteina monoclonal (IgM) • Carcinoma bronquial de renal • Neuromiopatía hipertiroidea • Polimiositis • Glucogenosis tipo II (Pompe) • Sífilis meningovascular, neuroborreliosis
8. Tratamiento (I): • Actualmente NO hay ningún tratamiento farmacológico eficaz para curar la enfermedad. • · Farmacológico: • Riluzol: acción antiglutamatérgica. Retrasa la evolución de algunas variedades de ELA. • Modelos experimentales combinan Rasagilina + Riluzol. • Memantina también es eficaz en animales de laboratorio. • 2 péptidos: Humanina y Colvelina han demostrado ser neuroprotectores en modelos experimentales de ELA en animales transgénicos. Podrían indicar una vía de tratamiento en la especia humana en el futuro.
Tratamiento (II): • Tratamiento sintomático: dietético, del insomnio, depresión, fallo respiratorio, etc. • Rehabilitación física: estiramientos, terapia ocupacional, dispositivos ortopédicos de ayuda. • Atención a las alteraciones del lenguaje: logopeda. • Cuidado nutricional: espesantes, gastrostomía. • Cuidado respiratorio: ventilación no invasiva, traqueotomía y ventilación invasiva.
Tratamiento (III): · Tratamiento MULTIDISCIPLINAR: Objetivo: conseguir mejorar el bienestar y la calidad de vida de los pacientes y sus familias. Componentes: • Neurológico • Rehabilitador • Respiratorio • Social • Soporte psicológico
9. Cuidados en el final de la vida: Con la progresión de la enfermedad, cambian las METAS del tratamiento a: control de los síntomas de disnea, dolor y sufrimiento. • Utilización de opiáceos: regla del “doble efecto”. • Retirada de tratamientos de soporte vital: derecho del paciente ético y legal. • Abstención voluntaria de comida y bebida: muerte en aproximadamente 1-3 semanas. • Sedación terminal: combinación de tratamiento sintomático agresivo y de retirada de tratamiento de soporte vital. • Suicidio asistido por el médico. • Eutanasia activa voluntaria.
Bibliografía: • Zarranz J.J, Neurología, Editorial Elsevier, 4ª edición, año 2008; 639-657, 883-885. • National Institute of Neurological Disorders and Stroke, Esclerosis lateral amiotrófica, Junio 2002. • AEBISCHER, Patrick y Ann C. KATO: «Combatir la esclerosis lateral amiotrófica», en revista Investigación y Ciencia, 376 (págs. 60-67), enero de 2008. • ESQUERDA COLELL, Josep E.: «Esclerosis lateral amiotrófica», en revista Mente y Cerebro, 17 (págs. 83-92), 2006. • Dávila González P., Torres Díaz C. V., Campos Pavón J., Ruiz Mateos B., Gandia González M. L., Truchuelo Díez M. T., Manual de Neurología y Neurocirugía, 3ª Edición, pág. 72