Download

1 / 26
270 likes | 502 Views
Winding Back the Evolutionary Clock. Biologists want to reconstruct the evolutionary history of genes, genomes, and speciesEvolutionary history helps us to understand the genomes we see todayPhylogenetic trees represent evolutionary relationships between species and are a vital ingredient in m

E N D
1. Uncovering evolutionary history: new methods for inferring phylogenies RSS Manchester
12 October 2005
2. Winding Back the Evolutionary Clock Biologists want to reconstruct the evolutionary history of genes, genomes, and species
Evolutionary history helps us to understand the genomes we see today
Phylogenetic trees represent evolutionary relationships between species and are a vital ingredient in many biological analyses
3. Today�s Talk An introduction to phylogeny
Maximum likelihood and distance-matrix frameworks
A new distance-matrix approach
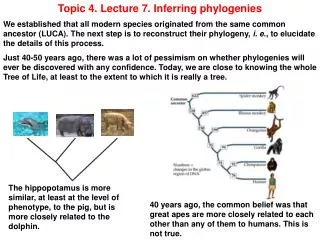
4. Evolutionary Trees Evolutionary relationships can be represented by trees, called phylogenies
Leaf nodes are extant species
Internal nodes are speciation events
Branch lengths show evolutionary distance
5. Biological Data
6. Sequence Evolution
7. Models of Nucleotide Substitution Model sites along the DNA string as evolving independently
Continuous time Markov chain with states A,C,G,T:
Define
Pij (t) = Prob (in state j at time t | given in state i at t = 0)
So that
P(t) = exp ?tQ
where
Q is the instantaneous rate matrix
? is the rate of mutation events, ?t represents branch length
Various models available for Q
8. Molecular Clocks Branch lengths represent evolutionary distance (typically number of nucleotide substitutions)
Rates of change may vary between branches
Molecular clock = no rate variation
9. Tree Likelihood Given a tree topology and branch lengths, evaluate the likelihood of the tree under the substitution model
10. Likelihood Maximization We can search for the maximum likelihood tree:
Pick an initial topology
Find the optimal set of branch lengths
Is this the highest likelihood we have seen?
Pick a new topology
11. Distance-Matrix Approaches Given a matrix of evolutionary distances, estimate the tree that gave rise to those distances
12. Comments Distance matrices
The distance matrix summarizes the information in the full sequence data set
Data loss � problematic for widely diverged sequences
Distance matrix is obtained from sequence data using a substitution model � many ways to do this
Comparison with likelihood
Distance matrix methods are less sophisticated...
... but they are much faster!
13. Least Squares Fitting Suppose we are given a tree topology and a distance matrix: how would we find branch lengths on the tree?
For two leaves i,j denote:
true distances on tree tij
observed distances dij
Assume that observed distances are unbiased estimates of the true distances:
Use branch lengths tij that minimize the error term:
14. Neighbour Joining Neighbour Joining (NJ) is defined by an agglomerative algorithm:
15. Comments NJ is hard to justify statistically...
... but it works surprisingly well!
Recent improvements to the algorithm have not introduced a thorough statistical framework
16. Our Methodology New distance-matrix method for constructing phylogenies
Motivated by the example of gene families � but also applies to species trees
Essential ingredients:
Distribution free, moment-based approach
Handles variance/covariance of distances more thoroughly than existing distance-matrix methods
17. Motivation: Families of Paralogs Certain genes have many copies within
the same genome
Examples: olfactory receptors, proteases, kinases
Appear to have evolved through duplications of individual genes, clusters of genes, and rearrangements within gene clusters
Phylogenetic tree for these genes ? history of gene duplication
Could we construct a more sophisticated history?
�Block duplications� of more than one gene
A history of linear arrangement along the genome
18. Assumptions (1) Molecular clock setting: necessary in order to consider events in which more than one gene is duplicated
In a block duplication, two or more genes are copied at the same time
The observed distances dij are the result of a random process perturbing the underlying true tree T
19. Assumptions (2) The observed distances dij are the result of a random process perturbing the underlying true tree T, that satisfies:
20. Building trees Adopt an agglomerative approach � �winding back the clock�
21. Scoring Joins (1) Suppose we have constructed T as far back as some time t. What is the covariance matrix for distances between nodes?
22. Scoring Joins (2)
23. Scoring Joins (3) Score tree using the goodness-of-fit of the calculated distances dt to expected distances:
Under suitable asymptotic assumptions this is a ?2 statistic
The distance vector dt is n x n so the covariance matrix is n2 x n2 and inverting it is potentially O(n6)
However, it can be inverted algebraically in O(n2) steps, and score evaluated in O(n) steps
24. Results Construction of large trees:
Comparison with other methods (NJ) in progress...
Issues
As a purely phylogenetic method it is held back by the molecular clock assumption
It is not a complete approach to inferring historical arrangements of paralogous genes, although it can incorporate duplication of more than one gene at a time
25. Conclusions Full probabilistic models for constructing phylogenies are unsuitable if there are many leaves
Existing distance-matrix methods could be improved upon
We have a new distance-matrix approach that improves upon standard approaches to variance / covariance
Future Work
Could we build our approach to covariance into a setting with no molecular clock?
Can we develop approaches that combine phylogenetic and arrangement information to build evolutionary histories?