Download

1 / 0
0 likes | 440 Views
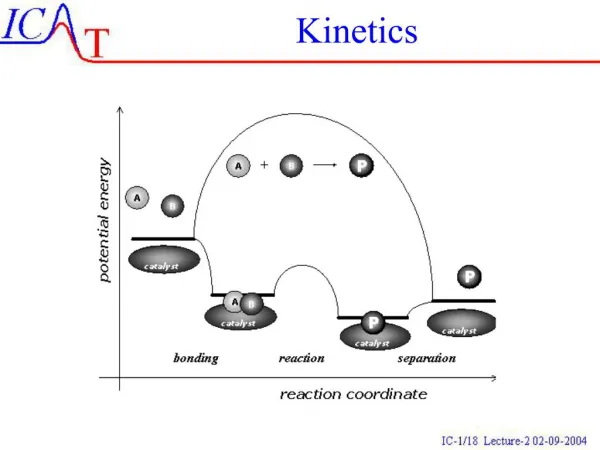
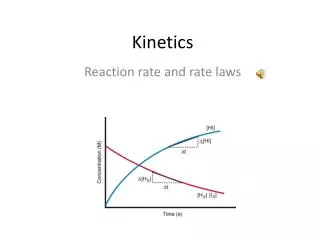
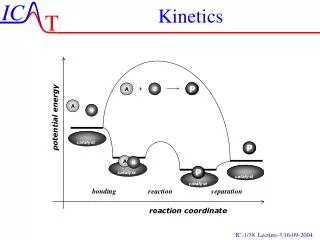
Kinetics. Ch 15. Kinetics. Thermodynamics and kinetics are not directly related Investigate the rest of the reaction coordinate Rate is important!. Chemical Kinetics. Kinetics – the study of the rates of chemical reactions

E N D