Download
1 / 2
20 likes | 184 Views
Hypercoagulability, portal vein thrombosis and liver fibrosis/cirrhosis in hereditary thrombophilia. Ming- Ching Shen 1,2* , Ching-Yeh Lin 1 , Shyuann-Yuh Lin 1 , Guan-Min Lai 1 , Shun-Sheng Wu 1 , Pei-Yuan Su 1 , Yueh -Ming Lin 1 1 Changhua Chirstinal Hospital, Changhua city, Taiwan

E N D
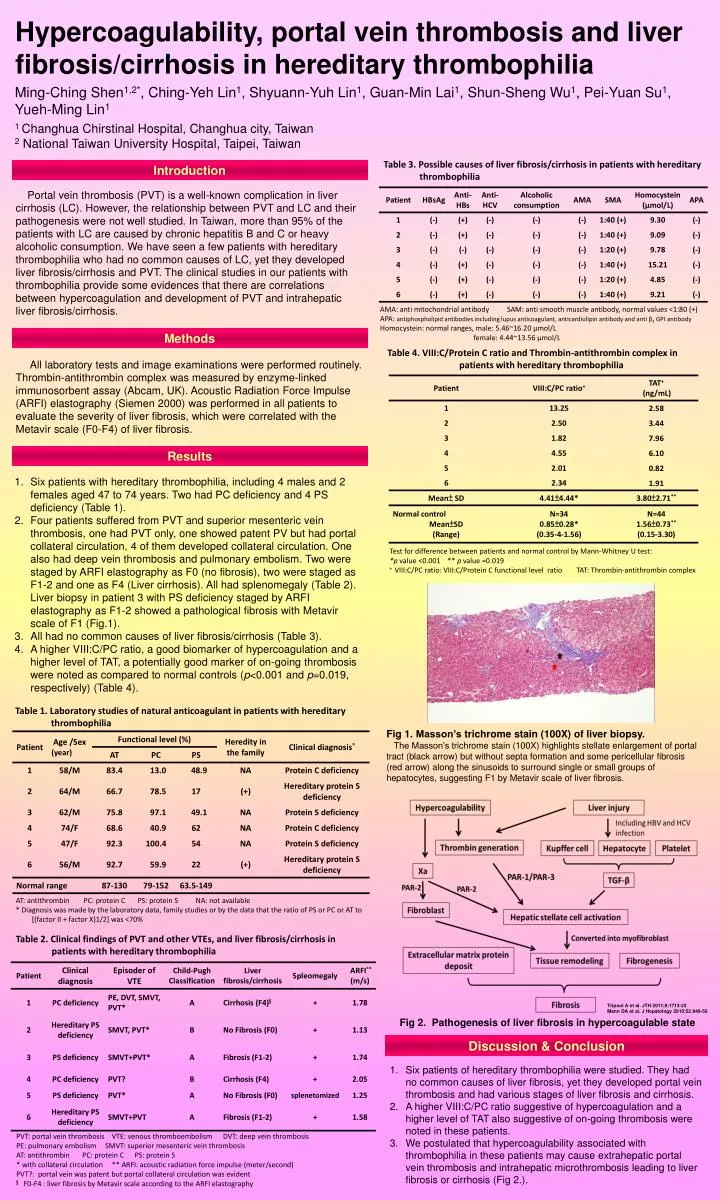
Hypercoagulability, portal vein thrombosis and liver fibrosis/cirrhosis in hereditary thrombophilia Ming-Ching Shen1,2*, Ching-Yeh Lin1, Shyuann-Yuh Lin1, Guan-Min Lai1, Shun-Sheng Wu1, Pei-Yuan Su1, Yueh-Ming Lin1 1 Changhua Chirstinal Hospital, Changhua city, Taiwan 2 National Taiwan University Hospital, Taipei, Taiwan Table 3. Possible causes of liver fibrosis/cirrhosis in patients with hereditary thrombophilia Introduction Portal vein thrombosis (PVT) is a well-known complication in liver cirrhosis (LC). However, the relationship between PVT and LC and their pathogenesis were not well studied. In Taiwan, more than 95% of the patients with LC are caused by chronic hepatitis B and C or heavy alcoholic consumption. We have seen a few patients with hereditary thrombophilia who had no common causes of LC, yet they developed liver fibrosis/cirrhosis and PVT. The clinical studies in our patients with thrombophilia provide some evidences that there are correlations between hypercoagulation and development of PVT and intrahepatic liver fibrosis/cirrhosis. AMA: anti mitochondrial antibody SAM: anti smooth muscle antibody, normal values <1:80 (+) APA: antiphospholipid antibodies including lupus anticoagulant, anticardiolipin antibody and anti β2 GPI antibody Homocystein: normal ranges, male: 5.46~16.20 μmol/L female: 4.44~13.56 μmol/L Methods Table 4. VIII:C/Protein C ratio and Thrombin-antithrombin complex in patients with hereditary thrombophilia All laboratory tests and image examinations were performed routinely. Thrombin-antithrombin complex was measured by enzyme-linked immunosorbent assay (Abcam, UK). Acoustic Radiation Force Impulse (ARFI) elastography (Siemen 2000) was performed in all patients to evaluate the severity of liver fibrosis, which were correlated with the Metavir scale (F0-F4) of liver fibrosis. Results • Six patients with hereditary thrombophilia, including 4 males and 2 females aged 47 to 74 years. Two had PC deficiency and 4 PS deficiency (Table 1). • Four patients suffered from PVT and superior mesenteric vein thrombosis, one had PVT only, one showed patent PV but had portal collateral circulation, 4 of them developed collateral circulation. One also had deep vein thrombosis and pulmonary embolism. Two were staged by ARFI elastography as F0 (no fibrosis), two were staged as F1-2 and one as F4 (Liver cirrhosis). All had splenomegaly (Table 2). Liver biopsy in patient 3 with PS deficiency staged by ARFI elastography as F1-2 showed a pathological fibrosis with Metavirscale of F1 (Fig.1). • All had no common causes of liver fibrosis/cirrhosis (Table 3). • A higher VIII:C/PC ratio, a good biomarker of hypercoagulation and a higher level of TAT, a potentially good marker of on-going thrombosis were noted as compared to normal controls (p<0.001 and p=0.019, respectively) (Table 4). Test for difference between patients and normal control by Mann-Whitney U test: *p value <0.001 ** p value =0.019 + VIII:C/PC ratio: VIII:C/Protein C functional level ratio TAT: Thrombin-antithrombin complex Table 1. Laboratory studies of natural anticoagulant in patients with hereditary thrombophilia Fig 1. Masson’s trichrome stain (100X) of liver biopsy. The Masson’s trichrome stain (100X) highlights stellate enlargement of portal tract (black arrow) but without septa formation and some pericellular fibrosis (red arrow) along the sinusoids to surround single or small groups of hepatocytes, suggesting F1 by Metavirscale of liver fibrosis. AT: antithrombin PC: protein C PS: protein S NA: not available * Diagnosis was made by the laboratory data, family studies or by the data that the ratio of PS or PC or AT to [(factor II + factor X)1/2] was <70% Table 2. Clinical findings of PVT and other VTEs, and liver fibrosis/cirrhosis in patients with hereditary thrombophilia Tripool A et al. JTH 2011;9:1713-23 Mann DA et al. J Hepatology 2010;52:949-50 Fig 2. Pathogenesis of liver fibrosis in hypercoagulable state Discussion & Conclusion Six patients of hereditary thrombophilia were studied. They had no common causes of liver fibrosis, yet they developed portal vein thrombosis and had various stages of liver fibrosis and cirrhosis. A higher VIII:C/PC ratio suggestive of hypercoagulation and a higher level of TAT also suggestive of on-going thrombosis were noted in these patients. We postulated that hypercoagulability associated with thrombophilia in these patients may cause extrahepatic portal vein thrombosis and intrahepatic microthrombosis leading to liver fibrosis or cirrhosis (Fig 2.). PVT: portal vein thrombosis VTE: venous thromboembolism DVT: deep vein thrombosis PE: pulmonary embolism SMVT: superior mesenteric vein thrombosis AT: antithrombin PC: protein C PS: protein S * with collateral circulation ** ARFI: acoustic radiation force impulse (meter/second) PVT?: portal vein was patent but portal collateral circulation was evident §F0-F4 : liver fibrosis by Metavirscale according to the ARFI elastography
Shang-Yi Huang*, WoeiTsay*, Ching-Yeh. Lin §, Shyuann-Yuh Lin§, Szu-Chun Hsu*and Ming-Ching Shen*§ * Department of Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan; Laboratory Medicine, National Taiwan University Hospital, Taipei, Taiwan; Haemophilia Center, National Taiwan University Hospital, Taipei, Taiwan § Department of Internal Medicine, Changhua Christian Hospital, Changhua, Taiwan (correspondence Ming-Ching Shen: 111710@cch.org.tw) TAIWAN INTRODUCTION Acquired haemophilia A (AHA) is a rare disorder caused by spontaneous development of autoantibody against FVIII coagulant protein (FVIIIi) in the non-hemophiliac population [1]. One main goal of therapy for AHA is to eliminate the FVIIIi [2]. The conventional immunosuppressive (IS) therapy consisting of corticosteroids alone or combined with cytotoxic agents (CTA; e.g., cyclophosphamide, azathioprine, or cyclosporine) has been used for such purpose [3]. Several emerging treatments, like rituximab-based (Rb) regimens or immune tolerance treatment (ITT) [3, 4] were reported to eliminate FVIIIi more efficiently. However, there is lack of predictors helpful for the choice between different treatments. Our aim here is to survey possible predictors of response to FVIIIi elimination therapy in our patients with AHA. PATIENTS & METHODS Between September 1987 and April 2010, a total of 46 Chinese patients with AHA who had the conventional IS treatment to eliminate FVIIIi in our institutes were retrospectively studied to identify potential predictors of the treatment response. A study of 46 patients with acquired haemophilia A in Taiwan finds elevated LDH level and mucosal bleedings associated with reduced response to immunosuppressive therapy RESULTS The demographics of these 46 patients are listed in Table 1. Notably four of them (8.7%) had active cancers. The median titer of FVIIIi was 22 Bethesda units (BU; range, 0.74–2414 BU) and 31 of them (67.4%) had FVIIIi titer more than 5 BU. Thirty-two patients (70%) had severe FVIII deficiency (FVIII:C < 1%), and the remaining 14 patients (30%) had residual FVIII:C detectable (median, 5.5%; range, 1.1–17). All other test results were generally within normal range except for hemoglobin (Hb) level (median, 9.7 gm/dl; range, 4.3–15.1 gm/dl), and lactate dehydrogenase (LDH) level (median, 506 IU/l; range, 203–1764 IU/l; reference range: 360–465 IU/l). At the median follow-up of 72 months, 21 of the 46 patients (45.7%) were dead. Causes of death were FVIIIi-related bleeding (n=5), sepsis (n=9), malignancy (n=5), and others (n=2). The flow chart on IS treatment to eliminate FVIIIi in our 46 patients was shown in Figure 1 Overall, after a median treatment duration of 15 weeks (range, 2–164 weeks), 29 patients (63%) had CR and the median time to CR was 16 weeks, which was comparable to other reports [1, 2, 3]. Interestingly, univariate analysis of predictors (including age > 65 years, FVIII:C < 1%, very high-titer FVIIIi > 50 BU, Hb < 8 gm/dl, white blood cell count < 4.0 x 109/l, platelet count < 1.5 x 1011/l, albumin < 3.5 gm/dl, LDH > 465 IU/l, APTT > 75 seconds, discrete bleeding symptoms, and whether associated conditions or not) of response to the IS therapy found an association of mucosal bleedings, very high-titer FVIIIi, and LDH > 465 IU/l with absence of a CR. Furthermore, by multivariate analysis, LDH > 465 IU/l (odds, 8.4 [95% CI, 1.4–51.1]; p=0.021) and mucosal bleedings (odds, 10.6 [95% CI, 1.8–63.9]; p=0.010), including gastrointestinal bleeding in 13 patients, urinary tract bleeding in seven patients, and intracranial bleeding in one patient, remained independent predictors of the absence of a CR. The CR rate in patients with both factors, one of these factors, and neither of these factors was 18%, 70%, and 92%, respectively, p=0.001. The positive predictive value of LDH > 465 IU/l for no CR to IS therapy was 55% and on contrary, the negative predictive value, terms of when LDH ≦ 465 IU/l there would be a CR to IS therapy, was 85%. Similarly, the positive and negative predictive value of mucosal bleedings for no CR to IS therapy was 57% and 80%, respectively. Table 1. Demographics of these 46 patients with aFVIIIi DISCUSSION & CONCLUSION To the best of our knowledge, this study of 46 patients with AHA is so far the largest case series of AHA in Chinese, and shows for the first time that elevated level of LDH (> 465 IU/l) and mucosal bleedings at diagnosis are potential factors associated with reduced response to the conventional IS therapy. We propose that mucosal bleedings (which occurred mostly in the gastrointestinal tract in our patients) might hamper absorption of oral prednisolone and CTA. However, the reason for association of elevated LDH with poor treatment response is not clear. Elevated LDH is a negative prognostic factor for some diseases, such as malignancies [5] and autoimmune disorders [6]. AHA results from circulating anti-FVIII autoantibody, which indicates the existence of autoreactive cell clones. In addition, an association of elevated LDH with sustained intramuscular bleeding in some cases of congenital haemophilia A has been reported [7]. Therefore, elevated LDH might reflect the severities and non-specific activities of conditions underlying this autoimmune phenomenon, no matter which was identifiable or not. To this point, it is noteworthy that the elevated level of LDH, though which was statistically independent by multivariate analyses in this study, could not be a totally independent nor direct factor from the clinical point of view. Carrying out a prospective study with enrollment of more patients to validate these findings is required. Nonetheless, since few clinical parameters can presently be used to guide AHA management [3], our findings may help clinical decision-making relating to the earlier implementation of emerging novel treatments such as Rb regimens or ITT protocols,instead of the conventional IS therapy, in patients with these negative predictors. On contrast, in AHA patients who do not have elevated level of LDH or mucosal bleedings at diagnosis, the conventional IS therapy may be feasible enough to eliminate FVIIIi. Table 2. Uni- and multi-variate analysis for the covariates to predict no response References 1. FranchiniM & Lippi G. Blood 2008; 112: 250–5. 2. Collins PW, et al Blood2007; 109: 1870–7. 3. Collins PW, et al. Br J Haematol 2009; 148: 183–94. 4. Zeitler H, et al. Haemophilia 2010;16: 95–101. 5. Duffy MJ. Ann ClinBiochem 2004; 41: 370–7. 6. Jain R, et al. Semin Arthritis Rheum 1994; 24: 173–82. 7. Forbes CD, et al. J ClinPathol 1972; 25: 1034–7.






