Download

1 / 27
270 likes | 476 Views
Pattern and string matching tools. Biology 162 Computational Genetics Todd Vision 9 Sep 2004. Some more pattern and string matching tools. Simple signatures Logos Position-specific Scoring Matrices PSI-BLAST Regular expressions Suffix trees. Sequence logos.

E N D
Pattern and string matching tools Biology 162 Computational Genetics Todd Vision 9 Sep 2004
Some more pattern and string matching tools • Simple signatures • Logos • Position-specific Scoring Matrices • PSI-BLAST • Regular expressions • Suffix trees
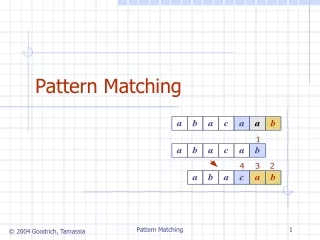
Sequence logos • Entropy of column j denoted Hj • Information content denoted Ij • How to draw a logo • Height of column given by Ij • Height of each symbol = fij x Ij
Information content • Information/Uncertainty is expressed in bits • There is a natural relationship to log base 2 • Imagine 64 shells, under one of which is a ball. • 6 guesses are required to find the ball • In this case, maximal uncertainty is log264=6 bits • In the case of 20 amino acids, maximal uncertainty is log220=4.32 bits.
Position-Specific Scoring Matrix • Constructed from conserved columns of a MSA • Log odds scores for each residue in each column, based on • Frequency of residue within column • Background frequency of residues • Takes advantage of the fact that columns differ in • Composition • Levels of conservation
Position Specific Scoring Matrix pos con A R N D C … A R N D C … Inf Pseu 1 M -1 -3 -3 -4 -1 … 0 0 0 0 0 … 0.50 0.16 2 W -3 -3 -4 -5 -3 … 0 0 0 0 0 … 2.32 0.26 3 I -1 -3 -2 -3 7 … 0 0 0 36 0 … 0.71 0.26 4 L -2 -3 -2 -3 -3 … 0 0 0 0 0 … 0.47 0.35 5 A 4 -2 -2 -2 -2 … 56 0 0 0 0 … 0.52 0.35 PSI-BLAST PSSM for DSCAM
Pseudocounts • If a residue is never seen in a particular column in of a MSA • What is the probability of ever seeing it there? • Not really zero… • Pseudocounts are added to actual counts to account for uncertaintly in column frequencies • Many methods • Laplace’s Rule • Add one to every count • Psudocounts grow less important as sample size gets large • Methods related to Bayesian priors - we will see later
Calculating scores in a PSSM • Sij is score for residue i at position j • xij is position-specific count of residue i • fi is background frequency of residue i • bij are pseudocounts • N sequences in alignment
PSI-BLAST • Can identify more distant homologs than possible via pairwise BLAST • Iterative BLAST • After 1st iteration, multiple alignment is computed for query and top matches • PSSM generated from alignment • PSSM used for subsequent iterations • PSSM refined each iteration
PSI-BLAST • Once high-scoring words are generated from PSSM, algorithm proceeds as before • Still very fast • l and K must be recalculated for each iteration
Regular Expressions (regex) • Can be thought of as a non-probabilistic rule for generating (or matching) a pattern • Used for • DNA/Protein signatures (e.g. Prosite) • Text parsing (e.g. in Perl)
Prosite regexes ID CBD_FUNGAL; PATTERN. AC PS00562; DT DEC-1991 (CREATED); NOV-1997 (DATA UPDATE); JUL-1998 (INFO UPDATE). DE Cellulose-binding domain, fungal type. PA C-G-G-x(4,7)-G-x(3)-C-x(5)-C-x(3,5)-[NHG]-x-[FYWM]-x(2)-Q-C In Perl regex syntax: CGG\w{4,7}G\w{3}C\w{5}C\w{3,5}[NHG]\w[FYWM]\w{2}QC In words: C followed by G followed by G followed by any 4 to 7 letters followed by G followed by any 3 letters followed by C followed by any 5 letters followed by C followed by an 3 to 5 letters followed by one of N, H or G, followed by any letter followed by one of F, Y, W, or M followed by any two letters followed by Q followed by C
Perl regex metacharacters • [ ] - character class (e.g. [abc] = a, b or c) • {min, max} - quantifiers • {exactly} • * - repetition, zero or more • + - repetition, one or more • ? - optional, zero or one • . - wildcard (any character) • ( ) - capture or delimit substrings • | - alternation (e.g. (a|b) = either a or b)
Regular expressions PatternMatches a[bc]d abd, acd ab{2,5}c abc, abbc, … abbbbbc ab*c ac, abc, abbc, … ab+c abc, abbc, … ab?c ac, abc a(bc|de) abc, ade
Regular expressions: limitations • Non-probabilistic: all matches match equally well • Hidden Markov models improve upon this • Cannot model dependencies among different positions • Neither can HMMs • For RNA matches, where dependencies matter, we need to allow more complex rules
Chomsky hierarchy of transformational grammars: a preview • General theory for modelling strings of symbols used in linguistics • Regular grammars • Context-free grammars • Context-sensitive grammars • Unrestricted grammars • Regular grammars (like regexes) are easy to parse, but are structurally limited • We will see context sensitive grammars for modelling RNA sequences
Suffix Trees • Data structure used for fast matching of sequence patterns • Helps to explain how BLAST can find word matches so fast • Commonly used for • Exact matching • Identifying repeated sequences
Suffix Trees • Rooted, directed tree for string S • |S| = m leaves, labeled 1..m • Edges labelled with substrings of S • Internal node has at most one edge for each symbol in alphabet • Concatenation of edge labels on path from root to leaf i equals suffix S[1..m]
root a ga tgac c tgac c tgac c 3 6 5 2 4 1 Suffix Trees: An Example S = ‘gatgac’
root a ga tgac c tgac c tgac c 3 6 5 2 4 1 Least common ancestor • LCA corresponds to shared prefix of suffix (e.g. path labeled ‘ga’ for nodes 1 and 4) • LCA can be retrieved in constant time
root a ga tgac c tgac c tgac c 3 6 5 2 4 1 If suffix trees are the answer, what is the question? • Rapid word matching • Find all occurrences of ‘ga’ in S = ‘gatgac’
If suffix trees are the answer, what is the question? • Longest common substring problem • Find the starting positions, length and identity of the longest substring that occurs in both S1 and S2 S1 = ‘gatgac’ S2 = ‘gatcac’ root t a ga c t t cac c cac cac c gac ac gac gac 3 3 6 6 4 5 5 2 2 4 1 1
If suffix trees are the answer, what is the question? • Find all direct palindromes (a substring concatenated with its reverse) in S=‘agattagct’ • Observation • Let Sr=‘tcgattaga’ • If a palindrome is centered between q and q+1 of S, then it is also centered between m-q and m-q+1 of Sr. • Solution • Construct joint suffix tree for S and Sr, find least common ancestor for all pairs q+1, n-q+1
Myriad uses for suffix trees • Direct and inverted repeats • Microsatellites • Transposons • Inverted palindromes • Restriction enzyme recognition sites • Imperfect matches • Algorithmic efficiency • Many efficient algorithms for traversing suffix trees • The trees themselves can be constructed in O(m) time
Reading assignment(for Tuesday and Thursday) • Durbin et al. (1998) pgs. 46-79 in Biological Sequence Analysis. • Markov chains • Hidden Markov models