Download

1 / 47
750 likes | 2.01k Views
Organometallic Chemistry. JHU Course 030.442 Prof. Kenneth D. Karlin Spring, 2009. Kenneth D. Karlin Department of Chemistry, Johns Hopkins University. karlin@jhu.edu http://www.jhu.edu/~chem/karlin/. p. 1.

E N D
Organometallic Chemistry JHU Course 030.442 Prof. Kenneth D. Karlin Spring, 2009 Kenneth D. Karlin Department of Chemistry, Johns Hopkins University karlin@jhu.eduhttp://www.jhu.edu/~chem/karlin/
p. 1 Organometallic Chemistry030.442 Prof. Kenneth D. Karlin Spring, 2009Class Meetings: TTh, 12:00 – 1:15 pm Textbook – The Organometallic Chemistry of the Transition Metals” 4th Ed., R. H. Crabtree Course Construction: Homeworks, Midterm Exams (1 or 2), Oral Presentations Rough SyllabusMost or all of these topics • Introduction, History of the field • Transition Metals, d-electrons • Bonding, 18 e–Rule (EAN Rule) • Ligand Types / Complexes • Types of Compounds M-carbonyls, M-alkyls/hydrides M-olefins/arenes M-carbenes (alkylidenes alkylidynes) Other • Reaction Types Oxidative Addition Reductive elimination Insertion – Elimination Nucleophilic/electrophilic Rxs. • Catalysis – Processes Wacker oxidation Monsanto acetic acid synthesis Hydroformylation Polymerization- Olefin metathesis Water gas-shift reaction Fischer-Tropsch reaction
p. 3 Reaction Examples • Oxidative Addition Reductive Elimination • Carbonyl Migratory Insertion • Reaction of Coordinated Ligands Vaska’s complex
p. 4 Reaction Examples - continued • Wacker Oxidation C2H4 (ethylene) + ½ O2 –––> CH3CH(O) (acetaldehyde) Pd catalyst, Cu (co-catalyst) • Monsanto Acetic Acid Synthesis CH3OH (methanol) + CO –––> CH3C(O)OH (acetic acid) (Rh catalyst) • Ziegler-Natta catalysts – Stereoregular polymerization of 1-alkenes (a-olefins) 1963 Nobel Prize Catalyst: Ti compounds and organometalllic Al compound (e.g., (C2H5)3Al ) • Olefin metathesis – variety of metal complexes 2005 Nobel Prize – Yves Chauvin, Robert H. Grubbs, Richard R. Schrock n CH2=CHR –––> –[CH2-CHR]n–
p. 5 Organo-transition Metal Chemistry History-Timeline • Main-group Organometallics 1760- Cacodyl – tetramethyldiarsine, from Co-mineral with arsenic 1899 –> 1912 Nobel Prize: Grignard reagents (RMgX) • 1827 – “Zeise’s salt” - K+ [(C2H4)PtCl3]– • n-Butyl-lithium Synthesis: PtCl4 + PtCl2 in EtOH, reflux, add KCl Bonding- Dewar-Chatt-Duncanson model
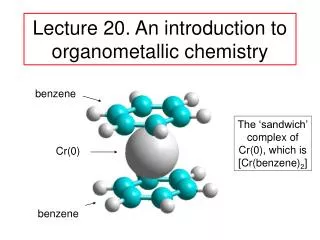
p. 6 Organo-transition Metal Chemistry History-Timeline (cont.) 1863 - 1st metal-carbonyl, [PtCl2(CO)2] 1890 – L. Mond, (impure) Ni + xs CO –––> Ni(CO)4(highly toxic) 1900– M catalysts; organic hydrogenation (---> food industry, margerine) 1930 – Lithium cuprates, Gilman regent, formally R2Cu–Li+ 1951 – Ferrocene discovered. 1952 -- Sandwich structure proposed Ferrocene was first prepared unintentionally. Pauson and Kealy, cyclopentadieny-MgBr and FeCl3 (goal was to prepare fulvalene) But, they obtained a light orange powder of "remarkable stability.”, later accorded to the aromatic character of Cp– groups. The sandwich compound structure was described later; this led to new metallocenes chemistry (1973 Nobel prize, Wilkinson & Fischer). The Fe atom is assigned to the +2 oxidation state (Mössbauer spectroscopy). The bonding nature in (Cp)2Fe allows the Cp rings to freely rotate, as observed by NMR spectroscopy and Scanning Tunneling Microscopy. ----> Fluxional behavior. (Note: Fe-C bond distances are 2.04 Å). Asides: Oxidation states US vs. UK 18-electrons ? (Cp)2Fe Cp = cyclopentadienyl anion) (h5-C5H5)2Fe (pentahapto) Solid-state structure
p. 7 Organo-transition Metal Chemistry History-Timeline (cont.) • 1955 - Cotton and Wilkinson (of the Text) discover organometallic-complex fluxional behavior (stereochemical non-rigidity) • The capability of a molecule to undergo fast and reversible intramolecular isomerization, the energy barrier to which is lower than that allowing for the preparative isolation of the individual isomers at room temperature. It is conventional to assign to the stereochemically non-rigid systems those compounds whose molecules rearrange rapidly enough to influence NMR line shapes at temperatures within the practical range (from –100 °C to +200 °C ) of experimentation. The energy barriers to thus defined rearrangements fall into the range of 5-20 kcal/mol (21-85 kJ/mol). Aside: Oxidation State 18-electron Rule
p. 8 Fluxional behavior; stereochemical non-rigidity(cont.) Butadiene iron-tricarbonyl • Xray- 2 CO’s equiv, one diff., If retained in solution, expect, 2:1 for 13-C NMR. But, see only 1 peak at RT. Cooling causes a change to the 2:1 ratio expected. • Two possible explanations: • Dissociation and re-association or (2) rotation of • the Fe(CO)3 moiety so that CO’s become equiv. • Former seems not right, because for example addition • of PPh3 does NOT result in substitution to give (diene)M(CO)2PPh3. • Note: You can substitute PPh3 for CO, but that requires • either high T or hv. So, the equivalency of the CO groups • is due to rotation without bond rupture, pseudorotation. 13C-NMR spectra CO region, only
p. 9 Berry Pseudorotation Pseudorotation: Ligands 2 and 3 move from axial to equatorial positions in the trigonal bipyramid whilst ligands 4 and 5 move from equatorial to axial positions. Ligand 1 does not move and acts as a pivot. At the midway point (transition state) ligands 2,3,4,5 are equivalent, forming the base of a square pyramid. The motion is equivalent to a 90° rotation about the M-L1 axis. Molecular examples could be PF5 or Fe(CO)5.
p. 10 The Berry mechanism, or Berry pseudorotation mechanism, is a type of vibration causing molecules of certain geometries to isomerize by exchanging the two axial ligands for two of the equatorial ones. It is the most widely accepted mechanism for pseudorotation. It most commonly occurs in trigonal bipyramidal molecules, such as PF5, though it can also occur in molecules with a square pyramidal geometry. The process of pseudorotation occurs when the two axial ligands close like a pair of scissors pushing their way in between two of the equatorial groups which scissor out to accommodate them. This forms a square based pyramid where the base is the four interchanging ligands and the tip is the pivot ligand, which has not moved. The two originally equatorial ligands then open out until they are 180 degrees apart, becoming axial groups perpendicular to where the axial groups were before the pseudorotation.
p. 11 Organo-transition Metal Chemistry History-Timeline (cont.) Methylmalonyl-CoA ––> Succinyl-CoA (CoA = coenzyme A) Catalysis of 1,2-shifts (mutases) or Homocysteine methylation 1961 – D. Hodgkin, X-ray structure – Coenzyme Vitamin B12(see other page) Oldest organometallic complex (because biological) (see other page) 1963 - Ziegler/Natta Nobel Prize, polymerization catalysts 1964 - Fischer, 1st Metal-carbene complex 1965 – Cyclobutadieneiron tricarbonyl, (C4H4)Fe(CO)3 – theory before experiment (C4H4) is anti-aromatic (4 p-electrons) With -Fe(CO)3, C4H4 behaves as aromatic 1965– Wilkinson hydrogenation catalyst, Rh(PPh3)3Cl 1971 – Monsanto Co. – Rh catalyzed acetic acid synthesis
Vitamin B-12 Co-enzyme p.12 Vitamin B-12 is a water soluble vitamin, one of the eight B vitamins. It is normally involved in the metabolism of every cell of the body, especially affecting DNA synthesis and regulation, but also fatty acid synthesis and energy production. Vitamin B-12 is the name for a class of chemically-related compounds, all of which have vitamin activity. It is structurally the most complicated vitamin. A common synthetic form of the vitamin, cyanocobalamin (R = CN), does not occur in nature, but is used in many pharmaceuticals, supplements and as food additive, due to its stability and lower cost. In the body it is converted to the physiological forms, methylcobalamin (R = CH3) and adenosylcobalamin, leaving behind the cyanide. 5-deoxyadenosyl group
p. 13 Organo-transition Metal Chemistry History-Timeline (cont.) 1973 – Commercial synthesis of L-Dopa (Parkinson’s drug) asymmetric catalytic hydrogenation 2001 Nobel Prize – catalytic asymmetric synthesis, W. S. Knowles (Monsanto Co.) R. Noyori,, (Nagoya, Japan), K. B. Sharpless (Scripps, USA) 1982, 1983 – Saturated hydrocarbon oxidative addition,including methane 1983 – Agostic interactions (structures)
p. 14 AGOSTIC INTERACTIONS: Agostic – derived from Greek word for "to hold on to oneself” C-H bond on a ligand that undergoes an interaction with the metal complexresembles the transition state of an oxidative addition or reductive elimination reaction. Detected by NMR spectroscopy, X-ray diffraction Compound above: Mo–H = 2.1 angstroms, IR bands were observed at 2704 and 2664 cm–1 and the agostic proton was observed at –3.8 ppm. The two hydrogens on the agostic methylene are rapidly switching between terminal and agostic on the NMR time scale.
p. 15 Organometallic Chemistry Definition:Definition of an organometallic compound Anything with M–R bond R = C, H (hydride) Metal (of course) Periodic Table – down & left electropositive element (easily loses electrons) NOT: • Complex which binds ligands via, N, O, S, other M-carboxylates, ethylenediamine, water • M–X where complex has organometallic behavior, reactivity patterns e.g., low-valent Oxidation State Charge left on central metal as the ligands are removed in their ‘usual’ closed shell configuration (examples to follow). d nfor compounds of transition elements N d < (N+1) s or (N+1) p in compounds e.g., 3 d < 4 s or 4 p
p. 16 d ncomputation – very important in transition metal chemistry d nzero oxidation state of M in M-complex has a configuration d n where n is the group #. Examples: Mo(CO)6 Mo(0) d n = d 6 (CO, neutral) HCo(CO)4 H is hydride, H–, --> --> Co(I), d n = d 8 Group 5 Group 6 Group 7 V(CO)6– Cr(CO)6 Mn(CO)6+ V(–1) Cr(0) Mn(+1) d 6d 6d 6 Isoelectronic and isostructural compounds (importance of d n) Effective Atomic # Rule; 18-Electron Rule (Noble gas formalism) # of electrons in next inert gas = # Metal valence electrons + s (sigma) electrons from ligands Rule: For diamagnetic (spin-paired) mononuclear complexes in organotransition metal compounds, one never exceeds the E.A.N.
p. 17 Cr(CO)6 Cr ---> d6 6 electrons (CO)6e– - pairs from 6 ligands 12 electrons ––> to [Ar] configuration 18 electrons (will see more in M.O. diagram) Consequence of EAN Rule: leads to prediction of maximum in coordination # Max coordination # = (18 – n) / 2 n is from d n . dn 10 8 6 4 2 0 Max Coord # 4 5 6 7 8 9 – Change in 2-electrons results in change of only one in Coord. # – Any Coord. # less than Max # ---> “coordinatively unsaturated” Fe(CO)42– Fe(CO)5 18 e– 18 e– Fe(–2) Fe(0) d 10d 8 4-coord 5-coord both Coord. Saturated
p. 18 [ReH9]2–e.g., as Ba2+ salt Re(VII), (Mn,Tc, Re triad) d 0, 9 hydride ligands; CN = 9 Geometry: Face capped trigonal prism A compound not obeying an rules Fe5(CO)15C Iron-carbonyl carbide
p. 19 Eighteen-Electron Rule - Examples Co(NH3)63+ Cr(CO)6 Obey 18-electron rule for different reasons Carbonyl Compounds in Metal-Metal Bonded Complexes less straightforward Fe2(CO)9 [p-Cp)Cr(CO)3]2 Co2(CO)8(2 isomers)
eg M+ M+ Δo t2g Free ion spherical six point charges spherically distributed octahedral ligand field M+ t2 M+ Δt e four point charges spherically distributed Free ion spherical tetrahedral ligand field p. 20 d6 Octahedral maximum of 6 coordinate
lower case letters for orbital dz2, dx2-y2 (e2g) (destabilized) 10Dq or Δo spherical field of 6 charges dxy, dxz, dyz (t2g) (stabilized) Oh p. 21 Picture of Octahedral Complex Various representations (ignore “s orbital”
p. 22 The five d-orbitals form a set of two bonding molecular orbitals (eg set with the dz2and the dx2-y2), and a set of three non-bonding orbitals (t2g set with the dxy, dxz, and the dyz orbitals). eg orbitals point at ligands (antibonding) appropriate symmetry for s-bonds to ligands s-bonds will be six d2sp3 hybrids ndz2, ndx2-y2, (n+1)s, (n+1)px,py,pz t2g orbital set left as non-bonding
p. 24 Standard MO diagram for Octahedral ML6 complexes with s-donor ligands e.g., [Co(NH3)6]3+ (18 e–) e.g., W(Me)6 (12 e–) Case I Electron-configuration unrelated to 18–-Rule 1st Row-Complexes with “weak ligands” Do small or relatively small, eg* only weakly antibonding No restriction on # of d-electrons –– 12 to 22 electrons
Case II Compounds which follow rule insofar as they never exceed the 18-e– rule • Metal in high oxidation state Do is large(r) (for a given ligand) radius is small –-> ligands approach closely ––> stronger bonding • 2nd or 3rd Row Metal - 4d, 5d Dois large(er) (for a given ligand); d-orbitals larger, more diffuse. Complexdn Total e–Complexdn Total e– ZrF62– 0 12 OsCl62– 4 16 ZrF73– 0 14 W(CN)83– 1 17 Zr(C2O4)44– 0 16 W(CN)64– 2 18 WCl6 0 12 PtF6 4 16 WCl6– 1 13 PtF6– 5 17 WCl62– 2 14 PtF62– 6 18 TcF62– 3 15 PtCl42– 8 16 Less than 18 e–, but rarely exceed 18 e– p. 26
p. 27 Similar Result if ligands are high in Spectrochemical Series e.g., CN–Do is larger V(CN)63– d2 Cr(CN)63– d3 Mn(CN)63– d4 Less than or equal to 6 d-electrons Fe(CN)63– d5 eg* not occupied Fe(CN)63– d6 Co(CN)63– d6 however Co(II) d7 ––> Co(CN)53– Ni(II) d8 ––> Ni(CN)42– and Ni(CN)53– Can have less than maximum # of non-bonding (t2g) electrons, because they are nonbonding. Addition or removal of e– has little effect on complex stability
p. 28 Docan get (or is) very small with p-donor ligands • F– example (could be Cl–, H2O, OH–, etc.) • Filled p-orbitals are the only orbitals capable of p-interactions • 1 lone pair used in s-bonding • Other lone pairs p-bond • The filled p-orbitals are lower in energy than the metal t2g set • Bonding Interaction • 3 new bonding MO’s filled by Fluorine electrons • 3 new antibonding MO’s form t2g* set contain d-electrons • Do is decreased (weak field) • Ligand to metal (L M) p-bonding • Weak field, p-donors: F, Cl, H2O • Favors high spin complexes
Metal Orbitals Molecular Orbitals Ligand Orbitals focus on this part only T1u both sets of d orbitals are driven ↑ in energy due to lower lying ligand orbitals 4p A1g 4s eg (σ*) Δo t2g (π*) Eg T1g,T2g T1u,T2u T2g 3d π-orbitals px, py t2g (π) A1g T1u Eg σ-orbital pz eg (σ) p. 29
antibonding eg (σ*) eg (σ*) eg (σ*) Δo Δo Δo t2g (π*) t2g (n.b.) both are antibonding t2g (π) non-bonding M-L bonding π-donor σ-donor π-acceptor smallest separation intermediate separation largest separation between sets of d-orbitals p. 30 Have discussed s-donor and p-donor – now p-acceptor
Molecular Orbitals Mo(CO)6 Metal Orbitals (only consider the d orbitals – 4s and 4p orbitals not included in the analysis) Ligand Orbitals t2g (π*) T1g, T2g T1u, T2u π* orbitals on CO (6 x 2 each - orthogonal) eg (σ* M-L) Eg Δo σorbitals on CO (6 x 1 each) T2g 4d A1g T1u Eg t2g (π) eg (σ M-L) p. 31 CASE III L high in spectrochemical series: CO, NO, CN–, PR3, CNR p-acid ligands – p-acceptors Can form strong p-bonds 18 e– rule followed rigorously Orbitals on M used in such p-bonding are just those which are non-bonding Result: Increase in Do Imperative to not Have electrons in eg* orbitals Want to maximize occupation of t2g because they are stabilizing
p. 33 Implications of 18e– Rule for Complexes with p-accepting ligands In octahedral geometry almost always have 6 d-electrons 12 electrons from ligands Other cases: # d-electrons and coordination # complementary • Coordination # exactly determined by electron-configuration and vice-versa (see previous notes) BrMn(CO)5(d ?) I2Fe(CO)4(d ?) Fe(CO)5(d ?) Ni(PF3)4(d ?) All 18-electron When M has odd electron ––––> metal-metal bond (often bridging CO’s) Mn2(CO)10 Co2(CO)8 Some 17 electron species known: V(CO)6d5 Mo(CO)2(diphos)2]+d5 See MO diagram: Want to fill stable MO’s’ there is a large gap to LUMO
dx2-y2 eg dxy Δo dz2 t2g dxz dyz (degenerate ) ML6 ML4 p. 34 Major Exception: d8 square-planar complexes As one goes across periodic table, d and p orbital energy Level splitting gets larger – hard to use p orbitals for s-bonding Common to have 4-coordinate SP complexes – dsp2 hybridization Which d-orbitals? Common for: Rh(I), Ir(I) Pd(II), Pt(II) Rationalize d-orbital splittings look at d-orbital pictures/axes
p. 36 Again, examples of complexes: dn C.N. Coord. Geom. Example(s) d10 4 Td Ni(CO)4, Cu(py)41+ d10 3 Trig.planar Pt(PPh3)3 d10 2 Linear (PPh3)AuX, Cu(py)2+ d8 5 TBP Fe(PF3)5 d8 4 (square) planar Rh(PPh3)2(CO)Cl (trans) d4 7 capped octahedral Mo(CO)5X2 d2 8 sq. antiprism ReH5(PMePh2)3, Mo(CN)84– d0 9 D3h symmetry [ReH9]2– tricapped trig. prism
p. 37 LIGANDS in Organometallic Chemistry: Ligands, charge, coordination # (i.e., denticity) X SnCl3 H (hydride) CH3 (alkyl, perfluoroalkyl) Ar RC(O) (acyl) R3E (E = P, As, Sb, N) R2P CO RNC (isonitrile, isocyanide) R2C (cabenoid, carbene) R2N N2 C2H4 (olefin, alkene) R2C2 (acetylene) C4H4 (cyclobutadiene) C3H5–(p-allyl) CH=CH-CH2– (s-allyl) benzene (arenes) p-C5H5 (p-Cp) p-C7H7 (tropylium) p-C3H3 (cyclopropenium, +) O (O-atom; oxide) NO (nitrosyl) ArN2+ (diazonium)
p. 38 Carbon Monoxide – exceedingly important ligand CO-derivatives known for all transition metals Structurally interesting, important industrially, catalytic Rxs Source of pure metal: Ni (Mond); Fe contaminated with Cu, purify via Fe(CO)5 Fe & Ni only metals that directly react with CO Source of oxygen in organics: RC(O)H, RC(O)OH, esters Processes: hydroformylation, MeOH ––> acetic acid double insertion into olefins, hydroquinone synthesis (acetylene + CO; Ru catalyst), acrylic acid synthesis (acetylene, CO, Ni catalyst) Fischer Tropsch Rx: CO + H2 ––> ––> CnH2n+2 + H2O Most of these involve CO “insertion”
p. 39 Metal-Carbonyl Synthesis: Reduction of available (in our O2-environment) metal salts, e.g., MX2, M’X3, other (e.g., carbonates) M-carbonyls generally in low-valent oxidation states ––––> “Reductive Carbonylation” Reductants: CO itself ( ––> CO2), H2, Na-dithionite Some Reactions: WMe6 + xs CO –––> W(CO)6 + 3 Me2CO NiO + H2(400 °C) + CO ––> Ni(CO)4 Re2O7 + xs CO ––> (OC)5Re–Re(CO)5 + 7 CO2 RhCl3 + CO + pressure + (Cu, Ag, Cd, Zn) –––> Rh4(CO)12 or Rh6(CO)16 Structures Possible: X-ray diffraction, Infrared spectroscopy Ni(CO)4 Fe(CO)5 M(CO)6 Td D3h Oh 2058 cm–1 2013, 2034 cm–1 2000 cm–1 Cl– acceptor/reductant
p. 40 H3B–CO = 2164 cm–1 no backbonding possible 13C NMR spectroscopy of M-CO fragments: 180 – 250 ppm Useful to use 13C enriched carbon monoxide Can be useful to observed “coupling” to other spin active nuclei, e.g., 103Rh or 13P
p. 41 Metal-Carbonyl Structures (cont.): Polynuclear Metal-Carbonyls
p. 45 The backbondingbetween the metal and the CO ligand, where the metal donates electron density to the CO ligand forms a dynamic synergismbetween the metal and ligand, which gives unusual stability to these compounds. Valence Bond formalism:
p. 46 C–O stretching frequencies, n(C-O) Put more electron density on metal – by charge – by ligands which cannot p-accept Remaining CO’s have to take up the charge (e–-density) on the metal See effects on n(C-O). Ni(CO)4 [Co(CO)4]– Fe(CO)42– 2057 cm–1 1886 cm–1 1786 cm–1 –––––––> –––––––> more –ve charge Mn(dien)(CO)3+ ~ 2020, 1900 cm–1 Cr(dien)(CO)3 ~1900, 1760 cm–1 (dien not p-acceptor)