Download
1 / 32
370 likes | 594 Views
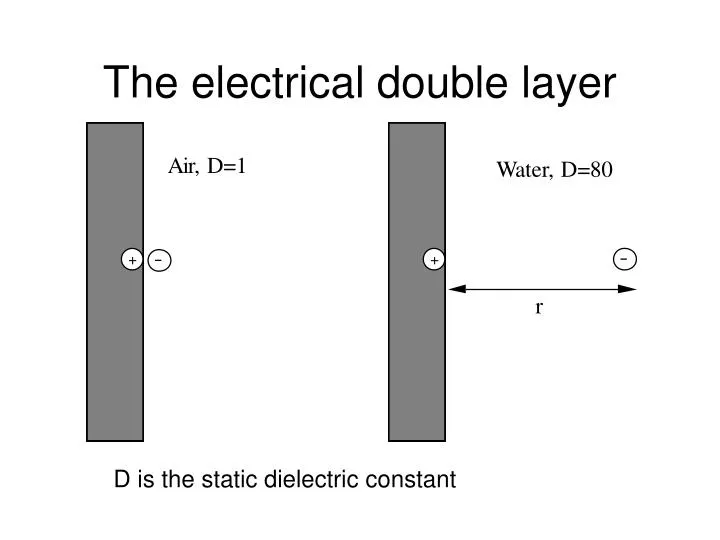
The electrical double layer. D is the static dielectric constant. From Coulombs equation,. By integration we can estimate the energy to separate a charge from the surface. In water W~1.9x10 -20 J, In Air W~1.5x10 -18 J Compare this with thermal energy 1 kT ~4.0x10 -21 J

E N D
The electrical double layer D is the static dielectric constant
From Coulombs equation, By integration we can estimate the energy to separate a charge from the surface. In water W~1.9x10-20 J, In Air W~1.5x10-18 J Compare this with thermal energy 1 kT ~4.0x10-21 J Clearly it is the high dielectric constant or polar nature of water which causes dissociation. In air or hexane (D~2), no dissociation is expected. This is why NaCl dissolves in water but not in oil.
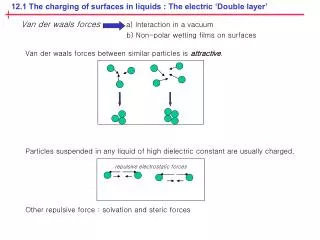
The real situation is more complicated as a large number of ions will be dissociated from each surface This generates a high electric field and a stronger attraction between the surface and the dissociated ions. Additionally, the solvated ions repel each other. In fact the dissociated ions do not leave the surface region completely. They form a “diffuse double layer”.
The charged surface and the diffuse ion layer of counterions form a double-layer (diffuse) capacitor.
Quantitative treatment of the electrical double layer is an extremely difficult problem. In order to treat the problem we make use of several assumptions and simplifications. Let us examine the case of an infinite, flat, charged planar surface. x is the distance normal to the surface. see Hunter, R Foundations of Colloid Science I & II, Oxford, 1989
Quantitative treatment of the electrical double layer is an extremely difficult problem. In order to treat the problem we make use of several assumptions and simplifications. x Let us examine the case of an infinite, flat, charged planar surface. x is the distance normal to the surface.
Y 0 Y x) ( 0 is the electric potential at the surface and (x) is the electric potential at a distance x from the surface
For a planar interface the potential is related to the charge density by Poisson’s equation (1) Where x is the net charge density in Cm-3 at distance x (ions per unit volume is equivalent to charge per unit volume).
In order to describe the decay of the potential from the surface we would like to determine x. The density (or population per unit volume) of any ion of charge Ziq must depend on it’s potential energy at that position. (Note Z is the valency). The potential energy is by definition given by Ziq(x). Note that q is the positive value of the electron charge, or the charge on a proton. Since any ion next to a charged surface must be in equilibrium with the corresponding ions in the bulk solution, it follows that the electrochemical potential of an ion at distance x from the surface must be equal to its bulk value. Thus:
where Ci(B) and Ci(x) are the ion concentrations in bulk and at distance x from the charged surface, and it is assumed that these are dilute solutions (ie ψ(B) = 0). This equation leads directly to the Boltzmann distribution, which can be used to obtain the concentration at any other electrostatic potential energy by the familiar relationship: In bulk solution we define the concentration of ion i as i(bulk). We would expect a Boltzmann distribution of ions determined by the ratio of the potential to the kinetic energies, therefore. (2) Note the sign of the ion charge is important
Why a Boltzmann Distribution? • Boltzmann’s law • the probability of finding molecules in a particular spatial arrangement decreases exponentially (negative sign) with the ratio of the potential energy of that arrangement compared with kT.
If the potential is positive then positively charged ions (co-ions) will be at a lower concentration near the surface than in the bulk. Ions of the opposite sign (counterions) will be at a higher concentration near the surface than in the bulk The net total charge density is given by (3) Assuming symmetric electrolytes (1:1, 2:2, 3:3 etc) and combining with equation (2) we obtain. (4)
This can be simplified to (5) Combining with equation (1) we obtain the Poisson-Boltzmann equation (6) After double integration and some algebra an exact analytical result is obtained, but if we assume low surface potential then equations become linear.
These equations can be greatly simplified by assuming low potentials. (ie values of (x)<25mV). The equation (6) reduces to ; (7) so (8) From inspection of equation (8) the physical meaning of the -1 (The Debye Length) is made clear. It is a measure of how the potential decays with distance from the surface. (9)
We now have a means of determining the ion profile from a surface and an approximate means for determining the potential profile for low potentials. We are also often interested in the charge density of the surface (Cm-2), which gives rise to the potentials. The total double layer must be electroneutral, therefore the charge at the surface 0must be equal and opposite in sign to the net charge of the diffuse layer D. (10) we obtain for a 1:1 electrolyte (using eqn 5), (11)
At low potentials equation (11) reduces to (12) The surface potential is therefore related to the surface charge density and the ionic composition of the solution.
Several assumptions have been made • The surface is flat, infinite and uniformly charged • The ions are assumed to be point charges, distributed according to the Boltzmann distribution • The solvent is represented solely by a dielectric constant • The electrolyte is assumed to be symmetrical
When two colloids interact We have already obtained a simple expression for the VdW’s interaction between two spherical particles. A simple result can be obtained for the interaction between diffuse electrical double layers, if we assume we are dealing with only low electrostatic potentials. This is known as the Debye-Huckel approximation. For simplicity we will first deal with two planar surfaces. From previous lectures recall that for low potentials the decay of the potential is described by: (1)
Recall that the Debye length is: • A natural length that the potential diminishes 1/e • Dependant only on the solution, not the surface charge
Non-interacting surfaces x>>-1 Interacting Surfaces x<-1 For interacting surfaces we apply the superposition approximation, which means the total potential at point x is given by the sum of the unperturbed potential from each surface x x =0 =m =0 =0
We therefore obtain for the potential at the midplane (2) At the midpoint the gradient of the potential is zero, d(x)/dx=0. Therefore no net electric field is acting on the ions at this point. However, at the midpoint there is a higher total concentration of ions than in the bulk. Osmotic pressure will then act to dilute the ions by drawing water into the region between the interacting surfaces. This is equal to the electrical double-layer repulsion between the charged plates. Hence, if we can determine the osmotic pressure at the mid-plane we can determine the repulsive pressure between the surfaces.
The osmotic pressure of an ideal solution is given by (3) where, is the solute concentration. The repulsive pressure is therefore given by the difference between the osmotic pressure at the mid-plane and the bulk solution and is given by: (4) where i refers to each ionic solute and and are the concentrations of each ionic solute in the mid-plane and in bulk solution, respectively.
As we know how the mid-plane potential varies with surface separation (equation 2), we can use the Boltzmann equation to calculate the concentrations of each ion at the mid-plane. By this procedure, assuming low potentials we obtain the result: (5) where the double-layer repulsive energy decays exponentially with distance and depends on the Debye length (ie electrolyte concentration) and the surface potential.
The interaction energy between two planar surfaces is obtained by integrating the osmotic pressure from infinity to x, which gives (6) By using a geometric factor the interaction energy between two spheres is obtained. (7) r r x (1) (2) (1) Again, the interaction decays with separation and depends on the surface potential and electrolyte concentration.