Download

1 / 47
480 likes | 670 Views
Approach to Inherited Neuropathies. Jodi Warman Chardon, MD, MSc Neuromuscular Fellow, McGill University Neurogenetics Fellow, University of Ottawa. Objectives. Develop approach to inherited neuropathies Appropriate history Relevant physical exam Review of electrodiagnostic testing

E N D
Approach to Inherited Neuropathies Jodi Warman Chardon, MD, MSc Neuromuscular Fellow, McGill University Neurogenetics Fellow, University of Ottawa
Objectives • Develop approach to inherited neuropathies • Appropriate history • Relevant physical exam • Review of electrodiagnostic testing • Develop appreciation for benefits and erroneous conclusions due to genetic testing • At the end, discussion of career or fellowship options if you would like • Please provide feedback: written or oral, positive and especially negative
Disclosures No disclosures
Quiz HMSN patients usually have primarily positive sensory symptoms at onset T/F With ROS on focused exam, it is necessary to review symptoms of visual changes, hearing loss, dysphagia and hoarseness T/F GJB1 is most common mutation causing inherited neuropathy T/F Patients with HMSN most often have decreased amplitudes and preserved conduction velocities and latencies on NCS T/F
Case X • 65 year old man, diabetic, smoker, alcoholic • 4 year history of sensory loss in his feet • Exam: MS, CN N • Motor: pescavus, distal weakness, length dependent • Sensory: distal pp decreased to ankles, vibration decreased to MM • Reflexes: 1+ symm reflexes except absent Achilles • What other information do you need?
Clinical approach to any neuropathy Alport and Sander, Continuum, 2012 Hx: Negative motor symptoms weakness, fatigue and wasting Positive motor symptoms include cramps, twitching and myokymia Negative sensory symptoms: hypesthesiaand gait abnormalities such as ataxia Positive sensory symptoms include burning or lancinating pain, paresthesias, “buzzing,” “tingling”
Clinical-assess for acquired causes • Exposures with • occupation (possibility of toxic exposures to solvents, glues, fertilizers, oils, and lubricants) • sexual history • recreational drug use • excessive alcohol intake • dietary habits • smoking Alport and Sander, Continuum, 2012
General PMHx (r/o acquired) Alport and Sander, Continuum, 2012 Medical (FHx) neuropathy focus: endocrinopathy(DM, hypothyroidism), renal insufficiency, hepatic dysfunction, CTD, and cancer. Surgical history: bariatric surgery, multiple orthopedic procedures, and multiple surgeries for ‘‘entrapped nerves’’ Medication list: (temporal) HAART, chemotherapy, Abx, herbal, etc ROS: rash/skin changes, arthralgias, dry eyes and mucous membranes, orthostasis, GI, and constitutional symptoms (fever, weight loss, night sweats).
Family History 3 Not just “no neuromuscular conditions…” unless clearly acquired cause Draw pedigree
Clinical-Clues to Inherited Neuropathy Siskind and Shy, Seminars in Neurology, 2011 Alportand Sander, Continuum, 2012 • Any symmetric, generalized polyneuropathy (+HNPP) • ….Family history • Symptoms less obvious to patient • Lack of positive sensory symptoms • Early age at onset, delayed milestones/ development • Ankle weakness (difficulty skating), stumble, running difficulties, balance difficulties vs later adult onset * • Associated skeletal abnormalities • Foot deformities • Scoliosis • Very slowly progressive • Lower, 10 years delay upper extremities (vs CMT1D)
Causes of Negative Family History Lawson & Gharibshahi, Seminars in Neurology, 2010 • Family data are frequently not available, incomplete, or unknown, a genetic cause is not ruled out: • initial sporadic mutation • expression can be variable • penetrance incomplete • late-onset disease • family members available for study may be pre-symptomatic • older relatives may have other comorbidities that confound the phenotype • older relatives may also pass away before evaluation
Pathophysiological Approach Suter and Scherer. Nature Reviews Neuroscience, 2003
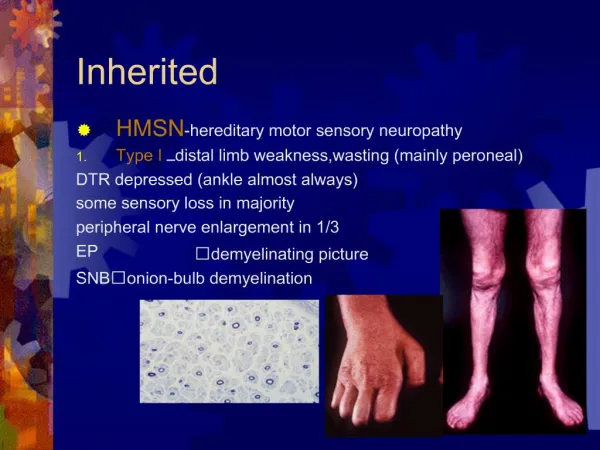
Neuropathy is the sole orprimary part of the disorder Reilly and Shy, J NeurolNeurosurgPsychiatry, 2009 Charcot–Marie–Tooth disease (CMT)= Hereditary Motor Sensory Neuropathy (HMSN) Hereditary neuropathy with liability to pressure palsies (HNPP) Hereditary sensory and autonomic neuropathies/hereditary sensory neuropathies (HSAN/HSN) Distal hereditary motor neuropathies (dHMN) Hereditary neuralgic amyotrophy (HNA)
Neuropathy part of a widespread neurological or multisystemdisorder Reilly and Shy, J NeurolNeurosurgPsychiatry, 2009 Familial amyloid polyneuropathy Disturbances of lipid metabolism Porphyrias Disorders with defective DNA Neuropathies associated with mitochondrial diseases Neuropathies associated with hereditary ataxias
CMT Choi et al. Human Mutation, 2012 • Charcot-Marie-Tooth disease (CMT) disease = Hereditary Motor Sensory Neuropathy (HMSN) • encompasses inherited neuropathies that demonstrate both genetic and phenotypic heterogeneity • i.e. PMP-22 [CMT1a, CMT1e, HNPP, early onset CMT (~CMT3 (previously known as Dejerine-Sottas) Congenital Hypomyelinating Neuropathy] • CMT results from mutations in more than 50 genes expressed in Schwann cells and neurons causing overlapping phenotypes
Complex CMT Genetics • 60% of all CMT=CMT1 • (80-90% is CMT1A with PMP 22 duplication> point mutation
CMT • “Classic”clinical phenotype: length-dependent degeneration characterized by • distal sensory loss and weakness • deep tendon reflex abnormalities • and skeletal deformities (foot, scoliosis) • Vs. “CMT-Plus” syndromes: • Optic atrophy, cataracts, glaucoma, deafness, dysphagia, respiratory, UMN etc
Electrodiagnostic studies (Classic) Shy M. Inherited Peripheral Neuropathies. Continuum. 2011 http://neuromuscular.wustl.edu/time/hmsn.html accessed September 15th, 2012
Current Organization of CMT Patzko and Shy, Continuum 2012 Shy M. Inherited Peripheral Neuropathies. Continuum. 2011 • CMT1=AD demyelinating <38 M/s • CMT2=AD axonal >38 M/s • CMT3=no longer used • Previously Dejerine-Sottas • CMT4=AR demyelinating or axonal • (Previously Refsumdisease, but now Refsum not considered part of CMT) • X-linked • Dominant Intermediate (meaning CV between 25-35 M/s
Approach to CMT Genetic Testing > 80% PMP22, MPZ, GJB1, MFN2 Siskind and Shy, Seminars in Neurology, 2011 Patzko and Shy, Continuum 2012
Approach to CMT Testing Siskind and Shy, Seminars in Neurology, 2011
Hereditary neuropathy with liability to pressure palsies (HNPP) Siskind and Shy, Seminars in Neurology, 2011 • Usually due to PMP 22 deletion (vs duplication with CMT1a) • Clinical features: • transient and recurrent motor and sensory mononeuropathies, usually at entrapment sites, such as the carpal tunnel, ulnar groove, and fibular head • duration: last hours, days or weeks or occasionally longer • HNPP can progress to long-term peripheral neuropathy phenotypically indistinguishable from CMT1
Hereditary sensory and autonomic neuropathies/hereditary sensory neuropathies (HSAN/HSN) • Distribution: Distal > proximal; Symmetric; Legs > Arms • Sensory Loss • Pain & Temperature (Small fiber) • Large fiber loss also occurs • Progressive • Spontaneous sensations • Lancinating pains, burning • Autonomic & reflex loss • Edx studies usually only useful later in disease www.neuromuscular.wustl.edu/sensory-small.html#hsan1 accessed Sept 13th, 2012
Distal hereditary motor neuropathies (dHMN) Rossor et al., Neuromuscular disorders, 2012 • Heterogeneous group of diseases with length-dependent predominantly motor neuropathy. • +/- minor sensory abnormalities and/or a significant upper-motor-neuron component, • often an overlap with CMT2 and with juvenile forms of ALS and HSP.
Why would you perform genetic testing in CMT? Lawson & Gharibshahi, Seminars in Neurology , 2010 Family planning and counselling Diagnostic certainty Relevance of unusual clinical features Avoidance of potential iatrogenic toxicities Scientific study
To choose no genetic testing Lawson & Gharibshahi, Seminars in Neurology 2010 Identification of a mutation in a presymptomaticor mildly affected individual Social stigmatization Selection of appropriate and rational testing Cost Genetic heterogeneity
Causes of Negative Genetic Testing Eccles, Practical Guide to Neurogenetics, 2008 Disease may be heterogeneous and the wrong gene has been tested Techniques used may be insufficiently sensitive to detect the causative mutation in that individual/family Mutation of unclear pathogenic significance may be found
Inheritance Pattern? 45 year old male with large fibre sensory loss, with myoclonus and hearing loss
Mitochondrial • Mitochondria are almost always inherited from the mother. • If a female has a mitochondrial trait, all of her offspring inherit it. • If a male has a mitochondrial trait, none of his offspring inherit it. • Complex phenotype, heteroplasmy • Mitochondrial inheritance not followed if nuclear mutations cause mitochondrial dysfunction
Inheritance Pattern? 12 year old female with with slowly progressive sensory loss in feet>hands with 5- weakness in l/e, palpable peroneal nerves Demyelination pattern in EDx
Autosomal Recessive Affected offspring are usually born to unaffected parents Appears in both sexes with equal frequency Trait tend to skip generations When both parents are hetrozygous, approx. 1/4 of the progeny will be affected Appears more frequently among the children of consanguinousmarriages
Inheritance Pattern? 5 year old boy with progressive, length dependent sensory and motor signs with NCS demonstrating demyelination
Autosomal Dominant Appears in both sexes with equal frequency Both sexes transmit the trait to their offspring Does not skip generations (but penetrance may not be 100% Affected offspring must have an affected parent unless they posses a new mutation When one parent is affected (het.) and the other parent is unaffected, approx. 1/2 of the offspring will be affected Unaffected parents do not transmit the trait
Inheritance Pattern? 18 year old boy with demyelination in S/M nerves on biopsy and progressive weakness and sensory loss; sisters and female cousins less affected
X-Linked Dominant • Both males and females are affected; often more females than males are affected • Does not skip generations. • Affected sons must have an affected mother; • Affected daughters must have either an affected mother or an affected father • Affected fathers will pass the trait on to all their daughters • Affected mothers if heterozygous will pass the trait on to 1/2 of their sons and 1/2 of their daughters
Inheritance Pattern? 30 year old male with 18 year history of progressive distal l/e weakness, now complaining of more hand weakness; mother has high arches and no weakness
X-Linked Recessive More males than females are affected Affected sons are usually born to unaffected mothers, thus the trait skips generations Approximately 1/2 of carrier mothers’ sons are affected It is never passed from father to son All daughters of affected fathers are carriers
Inheritance Pattern? • For the sake of completeness: • 30 year old male, thought he was in the urology clinic (mistake with neurology clinic by the clerk) due to infertility
Y-linked dominant Only males are affected It is passed from father to all sons It does not skip generations
Quiz HMSN patients usually have primarily positive sensory symptoms at onset F With ROS on focused exam, it is necessary to review symptoms of dysphagia and hoarseness, etcT GJB1 is most common mutation causing inherited neuropathy F Patients with HMSN most often have decreased amplitudes and preserved conduction velocities and latencies on NCS F
Summary Hereditary neuropathies can easily be misdiagnosed as acquired due to negative or incomplete family history Hereditary neuropathies exhibit both genetic and phenotypic heterogeneity Hereditary neuropathies have important consequences for prognosis, familial consequences (…and future molecular therapies)
Merci Questions? jwarman@cheo.on.ca