Download

1 / 36
420 likes | 1.14k Views
Chromatin Modification. Reading Seminar in Computational Biology Naomi Habib 5.1.2006. Chromatin Structure. Nucleosome (histone octamer wrapped with 146 bp of DNA). Chromatin Roles. Compactization Packing ~2 meters of DNA in to a ~10 micron diameter nucleus.

E N D
Chromatin Modification Reading Seminar in Computational Biology Naomi Habib 5.1.2006
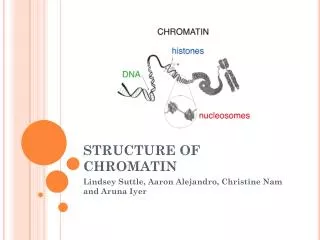
Chromatin Structure Nucleosome (histone octamer wrapped with 146 bp of DNA).
Chromatin Roles • Compactization • Packing ~2 meters of DNA in to a ~10 micron diameter nucleus. • Different compactization levels: euchromatin and heterochromatin. • Regulation of gene expression • Influencing DNA accessibility through Nucleosome Occupancy. • Histone variants. • Nucleosome tail modifications. • Co-regulation: Effect common chromatin regions. • Regulation of DNA replication, repair, recombination and more…
Nucleosome tail modifications • Lysine acetylations. • Histone Acetyl-Transferases (HAT) & Histone Deacetylases (HDAC). • Lysine and Argenine Metylations. • Modified by histone-metyl-transferase. • Phosphorilation. • Ubiquitination. • H2A ubiquitination affects 10-15% of this histone in most eukaryotic cells • ADP-ribosylation.
Possible regulation mechanisms • Histone modifications modulate the accessibility of DNA through structural changes of the chromatin (Horn and Peterson, 2002; Tse et al., 1998). • Modifications on different residues provide specific binding surfaces for transcription regulators. (Kurdistani and Grunstein, 2003; Corona et al., 2002; Deuring et al., 2000). • Histone code? number, variety, and interdependence of modifications. • Part of the process of protein signaling, promote switch-like behavior and ensure robustness of the signal(Schreiber and Bernstei,2002). • Epigenetic inheritance of information: histone modifications pattern inherited partially by the daughter cell(Jenuwein T & Allis C.D. 2001).
Articles Outline • Mapping Global Histone Acetylation Patterns to Gene ExpressionKurdistani,1 et al.Cell, June 2004. • Genome-wide Map of NucleosomeAcetylation and Methylation in YeastPokholok Et al. Cell, August 2005. • Genomic Maps and Comparative Analysis of Histone Modifications in Human and MouseBernstein et al. Cell January 2005.
Previous studies • Yeast nucleosomes occupancy is less dense in intergenic regions than ORF. (Lee et al., 2004). • Promotores and coding regions of transcribed genes had lower nucleosome occupancy (Lee et al., 2004). • Many works focusing on one or two Lysine residues or on a few genomic loci • Acetylation of Lysine residues on H3 and H4 – primarily associated with gene activation. (Grunstein,1997.Braunstein et at., 1993) • histone methylation associated with transcriptional activity or repression, depending on the specific residue (Zhang and Reinberg, 2001).
Previous studies (continued) • Genome wide analysis in D. melanogaster revealed parallel action of several ‘active’ modifications all linked to one another and to transcription levels(Schubeler D. et al, 2004) • Genome wide mapping of HATs and HDACs binding sites: (review by Bas Van Steensel, 2005). • HATs bind to all active promoters. • HDACs have a preferences for distinct gene classes
Mapping Global Histone Acetylation Patterns to Gene ExpressionSiavash K. Kurdistani,1 Saeed Tavazoie,2, Michael Grunstein1Cell, June 2004
Description of the work • Determining acetylation level of 11 Lysine residues in Saccharomyces cerevisiae. • Location analysis: Intergenic region & ORF arrays. • Normalizing the data for each array and variance-normalized across 11 sites arrays. • Clustered the data into clusters of modifications state. • Checked in each cluster: • Enrichment of co-expressed genes • Enrichment of genes from specific functional categories. • Search for unique cis DNA motifs. • Enrichment for binding of specific transcription factors.
Results • Lysines Acetylations are positively and negatively correlated. • H4K8 and K12 are strongly correlated in IGRs and ORFs. • Some significant differences between IGRs and ORFs. • hyper- and hypoacetylation lysines associated with transcription • hyperacetylation of histone H3K9/18/27 but hypoacetylation of H4K16 and H2BK11/16 are correlated with transcription.
Results (continued) • Acetylation patterns define groups of biologically related genes. • co-expressed, functional categories enrichment, Unique DNA motifs, Specific transcription factors binding. • Clusters distinguish groups with similar expression in one condition and differentially expressed in others. • TF Bdf1 binding is associated with acetylation (specifically H4K16). Model: Acetylation patterns used as surfaces for specific protein binding.
Genome-wide Map of NucleosomeAcetylation and Methylation in Yeast Dmitry K. Pokholok, Christopher T. Harbison, Stuart Levine, Megan Cole, Nancy M. Hannett, P. Alex Rolfe, Elizabeth Herbolsheimer,Julia Zeitlinger,Fran Lewitter, David K. Gifford,and Richard A. Young. Cell, August 2005
General Description • Genome-wide location analysis of nucleosome acetylation and methylation in Yeast. • Location analysis, array design: • Tile array of ~44,00 probes (85% of the genome). • 60-mer oligonucleotide, average probe density of 266 bp. • Improved resolution and accuracy. • Compared Histone-modifications-antibody data to a control with core-histone-antibody.
Checking Resolution -Gcn4 Location Analysis • Gcn4 TF of amino acid-biosynthetic genes. • FPR of 1% and FNR of ~25% over a set of 84 “positive” and 945 “negatives” genes. • Gene sets chosen according to location analysis data, DNA binding site motif and 2-fold change in mRNA levels dependent on Gcn4.
Nucleosome Occupancy • 20% reduction on average in histone occupancy in intergenic sequences relative to genic sequences. • No difference after normalization using control “no-antibody” data • 40% of promoters have difference from their gene.
Occupancy & Expression • Nucleosome occupancy inversely correlates with transcription. • Occupancy is reduced maximally at promoters of active genes. • In cells after oxidative stress, nucleosome occupancy dropped at genes know to be activated. Model: gene activation leads to reduced nucleosome density.
Histone Acetylation • Histone acetylation enriched at promoter regions and transcriptional start sites of active genes. • Acetylation at Gcn5 targets H3K9 and H3K14 and Esa1 targets H4 at Lys 5,8,12 and16. Model: transcriptional activators recruit Gcn5 and Esa1 to promoters of genes upon their Activation (Robert et al., 2004) .
Histone Acetylation • In general: strong correlation between acetylation of histone H3 and H4 (targeted by Gcn5 and Esa1) and transcriptional activity. • In contrast to Kurdistani et al. • After oxidative stress, histone acetylation increased at genes know to be activated and targeted by Gcn5 or Esa1.
Histone Metylation (H3K4me) • Previously shown: methyltransferase Set1 recruited to the 5’ end of actively transcribed genes targets H3K4. • H3K4me3 peaks at beginningof genes, with high correlation to transcription rates. • H3K4me2 is enriched in the middle of genes, and H3K4meat the end of genes.
Histone Metylation (H3K36me3) • Previously shown : H3K36 trimethylation targeted by Set2 (associated with the later stages of elongation). • H3K36me3 enriched throughout the coding region, peaking near the 3’ ends, correlated with transcription. model: Set2 is recruited by the elongation apparatus and that it methylates during active transcription.
Histone Metylation (H3K79me3) • Previously shown : The Dot1 histone methyltransferase modifies H3K79, within the core domain in 90% of all H3. • H3K79me3 enriched within the transcribed regions of genes with little correlation to transcription.
Genomic Maps and Comparative Analysis ofHistone Modifications in Human and Mouse Bradley E. Bernstein, Michael Kamal, Kerstin Lindblad-Toh, Stefan Bekiranov, Dione K. Bailey, Dana J. Huebert, Scott McMahon, Elinor K. Karlsson, Edward J. Kulbokas III, Thomas R. Gingeras, reStuart L. Schreiber, and Eric S. Lander. Cell, January 2005.
General Description • Maping the nonrepetitive portions of human chromosomes 21 and 22 for H3 Lys 4 di-,tri-methylation and Lys 9/14 acetylation. • Hepatoma cell line. • Compared lysine 4 dimethylation for several orthologous human and mouse loci. • cytokine cluster, IL-4 receptor region, and all four Hox clusters. • human and mouse primary fibroblasts. • Location analysis using tile arrays at 35 bp intervals. • Assessing results and threshold reliability using real time PCR.
Maps of Histone Modifications • Genomic maps that detail more than 39Mbp. • Modification sites cover 1% of the nonrepetitive portion of chromosome 21 and 22. • ChiP sample compared to whole genome DNA. • Relatively Uniform Histone Density across the Chromosomes. • no depletion on active promoters detected.
(Me2>>3) Di-,Tri-methylated H3K4 • Trimethyl Lys4 Correlate with 5’ Ends of active Genes. • 1 kb upstream to TSS of known genes / predicted genes / proximal to mRNA hybridization on the tile array. • 5’ end significantly enriched (6- to 8-fold) for polII Ser5-phosphorylated CTD. possible mechanism. • Dimethyl Lys4 Sites in the Vicinity of Active Genes and dependent on cell type. novel markers for cell state?
H3 Acetylation • H3 Acetylation Enriched at 5Ends of Genes and Strongly Correlates with Lys4 Methylation. • Consistent with previous studies. Systematic colocalization of methyl and acetyl marks reflect recruitment of complexes containing methylases and acetylases, and/or crosstalk between modifications.
Conservation Sequence identity cytokine and IL4R regions • Lys4-Methylated Sites Show Stronger Conservation of Locationthan Underlying Sequence. • Conserved 7-fold higher than expected at random.
Unique Profile for HOX Clusters • Broad Methylated Regions Overlay Substantial Portions of the Human and Mouse Hox Clusters • For example: 60kb region in HOXA with 85% K4me2.
HOXA cluster. HOX clusters (continued) • distinct expression patterns in other fibroblast lineage. • lineage-dependent active chromatin domains created at differentiation to maintain expression. • Detected transcription at 33% of methylated intergenic bases, ~10% in nonmethylated. -hybridizing RNA to the tiling arrays.
Summary • Nucleosome Occupancy • Human: Uniform & punctate Histone Density. • Yeast: Reduced histone occupancy in IGRs relative to ORFs. • Histone acetylation • Human: Enriched at 5’ Ends of Genes. • Yeast: Enriched at promoter regions and TSS of active genes. • Histone methylation • Human & Yeast: H3K4me3 peaks at 5’ Ends of active Genes. • Human H3K4me2in the Vicinity of Active Genes. / Yeast: H3K4me2 enriched in middle of genes • Yeast: Additional data regarding H3K4me3K79me3 and H3K36me3. • Conservation • Human & Mouse: H3K4me show stronger conservation of locationthan underlying sequence. • HOX cluster • Human & Mouse: Broad methylated lineage-dependent regions
Open questions • How are the acetylation patters established? • In IGRs may involve recruitment of specific sets of HATs and HDACs. • In ORFs determined by promoter regulatory motifs or association of HATs and HDACs with the RNAPII (elongating complex or Ser5-phosphorylated CTD). • What is the regulatory mechanism? Does the histone code exist? • ?