Download

1 / 23
240 likes | 261 Views
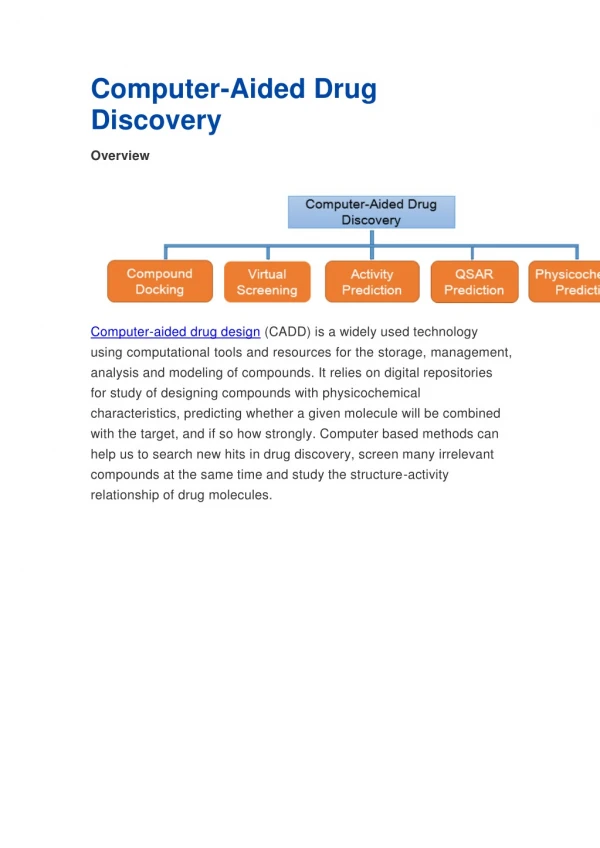
Computer Aided Drug Design. Hanoch Senderowitz Department of Chemistry Bar Ilan University. BIU-Valencia Workshop April 2010. Computer Aided Drug Discovery. Structure/ Sequence. Structure-based Modeling. Ligand-based Modeling. Known Ligands. Screening. Virtual Library. Scoring.

E N D
Computer Aided Drug Design Hanoch Senderowitz Department of Chemistry Bar Ilan University BIU-Valencia Workshop April 2010
Computer Aided Drug Discovery Structure/ Sequence Structure-based Modeling Ligand-based Modeling Known Ligands Screening Virtual Library Scoring Virtual Hits Binding Assays 3D Optimization In silico Leads Chemistry Biology Drug Candidate
Homology (Comparative) Modeling Given a sequence of amino acids, predict the 3D structure of the protein • Multiple sequence alignment • Multiple structure alignment Template selection • External servers • In-house tools Model generation • Energy minimization • Molecular dynamics • Virtual co-crystallization Model refinement • Model “health” • Agreement with available data • Enrichment experiments Model validation
In Silico Screening • Start: 2D representation of commercially available compounds • Filtration: Ligands and/or binding site characteristics • End: Multiple 3D conformations of ~100K compounds Library Generation Docking • Multiple docking tools BMA • Selection of the most plausible binding mode Scoring • Multiple scoring functions • Consensus scoring algorithms Selection • Selection of virtual hits • Biological assays
Donor Excluded volume Aromatic ring Aromatic ring Donor Shape based on largest active compound Acceptor Ligand-Based Screening • Pharnacophore: A 3D arrangement of function groups which is responsible for the biological activity • Obtained by the superposition of active (and inactive) compounds • A Database can be screened against pharmacophore
In Silico Screening Track Record (1) Conformational analysis; (2) IC50 from functionality assay; (3) Pharma collaboration; (4) Pharmacophore screening; (5) Ki estimated from IC50 OM Becker et al, PNAS101 (2004), 11304-11309
Airways Liver Pancreas Intestine Reproductive Tract Skin The Cystic Fibrosis Disease • CF is the most common lethal genetic disease among Caucasians • The number of CF patients is estimated at 70,000 worldwide, about 30,000 of which are in the US • In 2008, the median survival age of was ~37 years • CF results in pathologies in multiple organs • Depressed lung function, lung infection, inflammation and advanced lung disease • Currently, there is no cure for CF and the only treatment is symptomatic
The Molecular Basis of CF Normal lung • CF is caused by mutations to the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) which is the largest Cl- channel in the body • Most common disease causing mutation is DF508 • DF508-CFTR does not fold properly: Most channels does not reach the cell surface; those that do have impaired Cl- conductance • In absence of proper Cl- conductance the salt/water balance in the airways is disrupted leading to dehydration of the mucus layer lining the airways. • The dehydrated mucus layer becomes colonized by bacteria leading to chronic lung disease, lung failure and death • CFTR is a relevant target for developing CF therapeutics but its structure is unknown CF lung
wt-CFTR DF508-CFTR Model of Full Length Structure of CFTR • Site small and linear and aligned mainly by hydrophilic groups • Site sufficiently large for drug like compounds • Site supports specific interactions • Site is mostly linear and aligned by hydrophilic and aromatic moieties • Site sufficiently large for drug like compounds • Site supports specific interactions
~300 compounds from in silico screen YFP Fluorescence QuenchingFRT DF508 (rat) and A549 DF508 (human) pSAR FRT DF508Ussing chamberIn-house or at ChanTest In vitro Screening • Compounds tested in vitro in functional, electro-physiology assays in two cell lines • Assays measure channel conductance FRT Cells: 21 structure-based hits at 10 mM corresponds to a hit rate of 6.6% A549 Cells: 12 structure-based hits at 10 mM corresponds to a hit rate of 3.9% Similar screening campaigns reported in the literature yielded hit rates of 0.04-1.1% pSAR = Purchased SAR, i.e., purchasable analogs • Hits represent multiple scaffolds • In these assays, hits activity is similar to the best known CFTR corrector (Corr-4a) • Several hits show dual mechanism acting as both correctors and potentiators • Most promising hits entered lead optimization
CYP BBB Efficacy Solubility Permeability Binding hERG Lead Optimization: The Art of Balance
Binding • MM-GBSA simulations on a model system (Urokinase-type plasminogen activator (uPA)) • Good correlating when simulationinitiated from crystal structure (R2 = 0.75) • Poorer correlation when the binding mode could only be approximated (R2 = 0.60) • Poor correlation observed when only a model of the protein is available and /or when the binding mode is obtained through docking simulations • Challenges • Improved docking and scoring methods • Improved treatment of entropy
Astemizole (potent hERG binder) “Classic” hERG pharmacophore Privileged structures for e.g., GPCRs N+ When Binding is Improved… The hERG gene encodes a potassium channel conducting the repolarizing IKr current of the cardiac action potential. Drug related hERG inhibition could lead to a sudden cardiac death Binding to primary target often goes hand in hand with hERG binding Solution: hERG model
Permeability Affinity Hydrophobicity hERG binding When hERG is Reduced… • Due to the hydrophobic nature of the hERG binding site, increased polarity may reduce hERG binding. • Increased polarity will also lead to: • Increased solubility • Decreased permeation through biological membranes • Decreased permeation through the Blood Brain Barrier
Last But (Certainly) not Least Cyp inhibition may lead to toxicity via drug-drug interactions Cyp binding sites are large and promiscuous but are otherwise similar to “regular” binding sites CYP450-3A4 (PDB code 2v0m) Cavity size:950 Å3 to 2000 Å3 CYP450-2D6 (PDB code 2f9q) Cavity size:540 Å3
Optimization in Chemoinformatics and Drug Design • Drug Discovery is a multi-objective optimization problem • Successful drug candidates necessarily represent a compromise between numerous, sometimes competing objectives • Many other problems in chemoinformatics and drug design could be casted into the form of an optimization problem Synthesis design Docking & scoring QSAR/QSPR Optimization Engine Multiobjective QSAR Conformational search Classification Models Consensus scoring Diversity analysis
Energy Cartesian/internal coordinate 2 Cartesian/internal coordinate 1 The Target Function and Variables • Define a target function (f)and corresponding variables f = f(x1,x2,x3…xn) • Target function and variables related to the scientific problem • Target function and variables define a multi-dimensional surface
Random Move “Trial” DE Metropolis Test NO DE < 0 or exp(-DE/RT) > X[0,1] ? YES X[0,1] is a random number in the range 0 to 1 Tmax Temperature Temperature Temperature Temperature MC Tmin MC Steps MC Steps MC Steps MC Steps Monte Carlo/Simulated Annealing (MC/SA) Based Optimization Engine
Quantitative Structure Activity Relationship (QSAR) Quantitative Structure Property Relationship (QSPR) • Correlate specific biological activity for a set of compounds with their structure-derived molecular descriptors by means of a mathematical model • The nature of correlation, activity and descriptors are unlimited • BBB permeability = f (hydrophobicity, H-bonding potential) • Metabolic stability = f (presence/absence of specific fragments) • Protein crystallizability = f (amino acid composition, secondary structure)
Descriptors selection • Outliers removal • Generation of multiple models • Model(s) validation and selection • Consensus model • Validation • Predictions Descriptors Calculation Descriptors Selection Dataset Outlier Removal Internal Set External Set Multiple Divisions Y-Scrambling • Avoid chance correlation Training Set Test Set Model Derivation • Linear (MLR) • Non-linear (kNN) Model Selection Consensus Prediction • Average • SD QSAREngine
QSAR Model for Metabolic Stability in Human Liver Microsomes (HLM) Metabolism alters chemicals to speed their removal from the body and is performed primarily in the liver by the Cytochromes HLM experiments measure compounds resistance to metabolism Compounds incubated with HLM (vesicles containing drug-metabolizing enzymes) and their t1/2 half life determined
CYP Efficacy Permeability Binding BBB Solubility hERG The Grand Challenge • How can we reliably and consistently predict the pharmacological profile of bio-active compounds? • Basic scientific research • Practical applications in drug design • How can we make better drugs?
Acknowledgments • EPIX Pharmaceuticals • Lab members • Dr. Efrat Noy • Dr. Merav Fichman • Gal Fradin • Yocheved Beim • Funding • CFFT