Download

1 / 35
370 likes | 659 Views
Vasculitis Syndromes. Polymyalgia Rheumatica,Giant Cell Arteritis , Wegener’s Granulomatosis , Polyarteritis Nodosa. What is Vasculitis ?.

E N D
Vasculitis Syndromes PolymyalgiaRheumatica,Giant Cell Arteritis, Wegener’s Granulomatosis, PolyarteritisNodosa
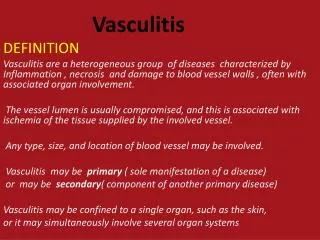
What is Vasculitis? • Disease characterized by inflammation of blood vessel walls, leading to altered blood flow through obstructed walls. This causes ischemia and tissue damage. • In addition there is an intense inflammatory rxn causing further systemic signs and symptoms • Can be fatal
You Should Suspect Vasculitis • Unexplained signs and sxs • Multisystem disease • Unexplained elevated ESR/CRP • Skin lesions (palpable purpura) • Ischemic vascular changes (Raynaud’s, gangrene, livedo, claudication) • Glomerulonephritis • Mononeuritis multiplex • Intestinal angina • Inflammatory ocular diease • Arthalgias/arthritis, myalgias • Sudden visual loss/headache
Select Vasculitides • PolymyalgiaRheumatica • Giant Cell or Temporal Arteritis • Wegener’s Granulamatosis • PolyarteritisNodosa
PolymyalgiaRheumatica (PMR) • Most ‘benign‘ of the group • Common: 50/100,000, age > 50, average age 75. Highest prevalence in northern European ancestry, females>>males • Cause unknown
PMR Clinical Presentation • Usually abrupt onset • Intense morning stiffness and pain that can last all day involving the shoulders and hip girdle area • No small joint involvement • Muscle strength normal • Fatigue and anorexia common • Elevated CRP and ESR; anemia of chronic disease, elevated platelets • 15% get GCA (more later)
PMR Treatment • Low Dose Steroids (10-20 mg/day) • The only drug that works • Look to normalize the CRP and ESR; if they continue to be elevated, rethink the dx (?paraneoplastic syndrome or GCA) • Usually self-limited: 65% of patients able to taper off Prednisone by 1 year, >85% in 2 years • Disease flares not uncommon as prednisone is tapered and may require dose adjustments
Giant Cell Arteritis • Can occur exclusively but often seen with PMR • Rare: 15/100,000 • Age >50 • Cause unknown • Involves the medium/large blood vessels of the head and neck including the blood vessels that supply the optic nerve
GCA Pathophysiology • Unknown trigger causes inflammatory response with the release of IL-1 and IL-6. • This leads to systemic symptoms and the infiltration of inflammatory cells into the adventitia of the temporal and other involved arteries • Typical histologic pattern: Giant Cells
GCA Clinical Presentation • Variable • Scalp tenderness • Temporal headaches • Jaw Claudication • Sudden loss of vision • +/- PMR sxs • Rare- upper extremity claudication due to subclavian involvement • Constitutional sxs: FUO, wt loss, fatigue • Bounding OR absent temporal artery pulses • Rarely subclavian bruits
GCA Diagnostic Studies • Temporal Artery Biopsy is the gold standard • Elevated ESR and CRP, usually levels higher than in PMR • Anemia • Elevated LFTs not uncommon
Treatment of GCA • High dose Steroids (60 mg/day) is the only drug that works • Slow taper over time usually 1-2 years. Some patients require low dose (<10 mg/day) chronically
GCA Complications • Blindness • Scalp Necrosis • Lingual Infarction • Aortic Dissection/Aneurysm • Complications from high dose steroids: osteoporosis, cataracts, elevated blood sugars, wt. gain etc.
Wegener’s Granulomatosis (WG) • Potentially fatal vasculitis involving small vessels • Rare: 3-14/million, more common in whites, any age but rare in children • Pathology shows necrotizing granulomas usually in upper airways, lungs and kidneys
WG Pathophysiology • Complex immunopathogenic events in which the production and activity of ANCAs (usually c-ANCA) play a central role. These autoantibodies interact with primed neutrophils to cause vascular injury and necrosis. • Histologic lesions show granulomas
WG Clinical Presentation • Variable, multisystem involvement • Organs: • Eyes: episcleritis/scleritis, proptosis due retro-orbital mass • CNS: rare mass lesion • Upper airway: otitis media, nasal chondritis, sinusitis with purulent drainage and epistaxis, ulcerations, subglotticstenosis • Kidney: neprotic syndrome, proteinuria, renal failure • Skin: palpable purpura due to leukocytoclasticvasculitis, pyodermagangrenosum, panniculitis • Lung: cough, hemoptysis, hemorrhage, resp failure • Cardiac: pericarditis, conduction abnormalities • Systemic: fever, night sweats, wt loss, fatigue
WG Diagnostic Studies • Presence of c-ANCA (cytoplasmic staining pattern antineutrophilcytoplasmic antibodies + clinical picture is often enough to make the diagnosis. It is + 80-90% of generalized WG. • If the c-ANCA is -, tissue biopsy of lung or kidney is recommended. • “Limited” refers to disease limited to the airways; c-ANCA often is -.
Additional labs • Elevated CRP and ESR • Anemia, leukocytosis, & thrombocytosis • Elevated Cr • Active urine sediment with red cell casts, hematuria and proteinuria
WG Clinical Course/Progression • Prior to immunosuppression therapies, WG was uniformly fatal. Now survival rates almost 90% with aggressive treatment. • High dose steroids and Cyclophosphamide are cornerstone of therapy. Methotrexate or Azathioprine sometimes used as steroid sparing agents.
PolyarteritisNodosa (PAN) • Medium vessel vasculitis • Can be caused by Hep B • 5/million cases • Peak incidence 50’s & 60’s, slightly more common in males
PAN Pathophysiology • In HepB assoc cases immune complexes play significant role • In non Hep B cases, the pathophysiology is less understood
PAN Clinical Presentation • Systemic: fever, fatigue, wt loss • Abdominal pain due to mesenteric angina/ischemia • Mononeuritis multiplex • Myalgias/arthalgias/mild arthritis • Hypertension • Skin: livedoreticularis, palpable purpura, fingertip ulceration, subcutaneous nodules • Testicular pain or tenderness
Complications of PAN • Chronic renal failure • Bowel perforation • Stroke/cerebral hemorrhage due to HTN • Foot/wrist drop
Labs of PAN • Elevation of acute phase reactants (ESR, CRP etc) • Absence of ANCA • Elevated transaminases, decreased albumin • +/- Hep B • Urine: proteinuria and hematuria without casts
Imaging Studies of PAN • Mesenteric and/or renal angiography is the test of choice • Biopsies seldom done
PAN Treatment • High dose steroids and Cyclophosphamide • Methotrexate or Azathioprine is used as steroid sparing agents later once the disease is controlled • Treatment for Hep B with antivirals. Sometimes plasma exchange is used to remove immune complexes