Download
1 / 53
560 likes | 2.31k Views
12 章 アルケンの反応. 12 -1 付加反応はなぜ進行するのか:熱力学的考察. 発熱反応. 表 12 ー 1. Δ H ゜: –11 ~ –33 kcal/mol. アルケンに対する付加反応はエネルギーの放出を伴って進行し, まちがいなく付加生成物を与えるはずである。. Δ H °= –33 kcal/mol. 101. 101. 12-2 触媒を用いる水素化反応. 長時間変化なし. 室温で反応進行. CH 3 CH 3. H ー H. H 2 C=CH 2. - H ー H. - H 2 C=CH 2. 触媒表面.

E N D
12-1 付加反応はなぜ進行するのか:熱力学的考察12-1 付加反応はなぜ進行するのか:熱力学的考察 発熱反応 表12ー1 ΔH゜: –11 ~ –33 kcal/mol アルケンに対する付加反応はエネルギーの放出を伴って進行し, まちがいなく付加生成物を与えるはずである。 ΔH°= –33 kcal/mol 101 101
12-2触媒を用いる水素化反応 長時間変化なし 室温で反応進行 CH3CH3
HーH H2C=CH2 - HーH - H2C=CH2 触媒表面
12-3p結合の求核的性質:ハロゲン化水素の求電子的付加12-3p結合の求核的性質:ハロゲン化水素の求電子的付加 求電子的攻撃 求核的攻撃 カルボカチオンの生成
Markovnikov則:ハロゲン化水素のプロトンは置換基のより少ない炭素にMarkovnikov則:ハロゲン化水素のプロトンは置換基のより少ない炭素に 結合する=生成するカルボカチオンの相対的安定性 置換基がより少ない
Markovnikov則は生成するカチオンの相対的安定性を考えて理解するMarkovnikov則は生成するカチオンの相対的安定性を考えて理解する プロペンのC2でのプロトン化 遷移状態1 第一級カルボカチオン (観測されない) プロペンのC1でのプロトン化 遷移状態2 第二級カルボカチオン (有利である)
プロペンのC1及びC2でのプロトン化:ポテンシャルエネルギー図プロペンのC1及びC2でのプロトン化:ポテンシャルエネルギー図 TS-1 第一級カチオン (観測されない) TS-1 E 有利である 反応座標 非対称アルケンに対するHXの付加反応では,プロトン化が第一段階として起こるが, このときより安定なカルボカチオンが生成するようにプロトンの付加が進行する。
求電子付加反応はカルボカチオンの転位を伴うことがある求電子付加反応はカルボカチオンの転位を伴うことがある カルボカチオンの 転位を伴った生成物 通常のMarkovnikov 付加による生成物 求核性が低い 第一段階 競争反応
12-4求電子水和反応によるアルコール合成:熱力学支配12-4求電子水和反応によるアルコール合成:熱力学支配 Markovnikov 則 Electrophilic hydration 全ての段階が可逆である 機構 SN1 E1
アルケンのプロトン化が可逆的であるためアルケンの平衡が達成されるアルケンのプロトン化が可逆的であるためアルケンの平衡が達成される =アルケンの可逆的プロトン化 H+(-H2O) - H+ H+ - H+ H+ H+ 触媒量の酸 触媒量の酸 シス トランス ★ 異性体間での相互変換=interconversion) ★ 熱力学支配(= thermodynamic control)による反応例
12-5アルケンに対するハロゲンの求電子付加反応12-5アルケンに対するハロゲンの求電子付加反応 アルケンのハロゲン化 , CCl4 室温あるいは 室温以下の温度 1-hexene 90% 1,2-dibromohexane アルケンに臭素を加えると赤茶色をした臭素の色は即座に消える Please remember ! 飽和化合物と臭素の反応はラジカル機構で進む反応 で,その反応速度はずっと遅く,光や熱で反応を開始させる必要がある。
臭素化反応はアンチ付加で進行する (1R,2R) (1S,2S) 83% yield cyclohexene racemic trans-1,2-dibromocyclohexane 2-ブテンの立体特異的な臭素化 racemic cis-2-butene (2R,3R) (2S,3S) 2,3-dibromobutane meso trans-2-butene (2R,3S) (2S,3R)
環状ブロモニウムイオンによって立体化学が説明できる環状ブロモニウムイオンによって立体化学が説明できる Bromonium ion (ブロモニウムイオン) ブロモニウムイオンの生 成の分子軌道による表現 脱離基 SN2 + 求核剤
Observing Halonium Ions: X-ray Crystallography Bellucci, Lenoir, Herges, J. Am. Chem. Soc. 1995 Lenoir, Chiappe, Chem. Eur. J. 2003 -SbCl6 SbCl5, Cl2 Kochi, Chem. Commun. 1998
12-6求電子付加反応の一般性 ブロモニウムイオンは臭化物イオン以外の求核剤によっても捕捉される - H+ - Br- 分子内 Williamson ether synthesis
ハロニウムイオンの開環反応は位置選択的であるハロニウムイオンの開環反応は位置選択的である ここにより大きな部分陽電荷 SN2 ブロモニウムイオンの置換基のより多い炭素を攻撃する 1-bromo-2-methyl-2-propanol オキサシクロプロパンの酸触媒下での求核的開環反応と 非常によく似ている(390ページを見よ)
12-7オキシ水銀化-脱水銀化:特殊な求電子付加反応12-7オキシ水銀化-脱水銀化:特殊な求電子付加反応
12-8ヒドロホウ素化-酸化:立体特異的逆 Markovnikov 水和反応 ヒドロホウ素化 Hydroboration: H. C. Brown 1979 Nobel Prize ヒドロホウ素化の機構 d+ d+ d- 空のp軌道
‡ ボラン-アルケン錯体 反応剤 位置選択性:立体的要因 ヒドロホウ素化・酸化
アルキルボランの酸化の機構 合成目的の アルコール (RO)3B + 3 NaOH Na3BO3+ 3 ROH
12-9 ジアゾメタン:カルベンとシクロプロパンの合成12-9 ジアゾメタン:カルベンとシクロプロパンの合成 ジアゾメタンからメチレンが生成し,メチレンはアルケンをシクロプロパンに変換する
ハロゲン化されたカルベン Simmons–Smith反応剤 ジヨードメタン Simmons–Smith反応剤 (カルベノイド)
12-10オキサシクロプロパン(エポキシド)の合成:過酸によるエポキシ化反応12-10オキサシクロプロパン(エポキシド)の合成:過酸によるエポキシ化反応 過 酸 (peracid)
cis-体:生成しない オキサシクロプロパン生成の相対速度 ・エポキシ化の位置選択性 アルキル置換基の数が 増すほど増大する
オキサシクロプロパンの生成機構 求電子的酸素 遷移状態 右90°回転 90° nーp*軌道相互作用:安定化 olefin plane Sarzi-Amadè et al. J. Org. Chem. Dec. 2002
anti dihydroxylation(アンチジヒドロキシル化) プロトン化 SN2
12-11四酸化オスミウムによる隣接シンジヒドロキシル化12-11四酸化オスミウムによる隣接シンジヒドロキシル化 四酸化オスミウムによるアルケンの酸化の機構 オスミン酸エステル 三つの電子対が同時に移動してOs(VI)を含む環状の エステルを与える協奏的付加反応
還元 OsO4, THF25 ℃, 48 h H2S OsO4: 高価,毒性強 RCOOH 過剰酸化 濃紫色: 反応が終 れば無色 褐色沈殿 色の変化はオレフィンの存在の確認
エナンチオ選択的ジヒドロキシ化反応の抗腫瘍剤合成への応用エナンチオ選択的ジヒドロキシ化反応の抗腫瘍剤合成への応用 ハイライト●12-3
12-12酸化的開裂反応:オゾン分解 =オゾン(ozone)
12-13ラジカル付加反応: 逆 Markovnikov 付加体の生成 ラジカル連鎖反応機構 全く異なる二つの結果が報告され,アルケンの化学に混乱が! 1930年代M. S. KharaschによってROORの存在が明らに!
DH°= + 39 kcal/mol DH°= - 17 kcal/mol 104 kcal/mol DH°= - 5 kcal/mol 98.5 kcal/mol DH°= - 11.5 kcal/mol HCl やHI の付加では伝播 段階の一つが吸熱反応なの で,反応が非常に遅くなり 連鎖反応が停止(イオン反 応は可) 結合解離エネルギー(kcal/mol): HーCl: 103 C=C: 65CーCl: 84 HーBr: 87CーBr: 71 HーI: 71CーI: 56
伝播段階 1: DH°= -23 kcal/mol 伝播段階 2:DH°= +3 kcal/mol DH°= -17.5 kcal/mol SーH: 81 kcal/mol SーC: 62 kcal/mol Dibenzoyl peroxide ジベンゾイルペルオキシド Bis(1,1-dimethylethyl) peroxide ビス(1,1-ジメチルエチル)ペルオキシド
12-14アルケンの2量化,オリゴマー化ならびに重合12-14アルケンの2量化,オリゴマー化ならびに重合 重 合 カルボカチオンはπ結合を攻撃 H+ ~H+ ~H+ 2量化
合成高分子の大まかな歴史 1856 Alexander Parke: セルロイド = ニトロセルロース + ショウノウ ● 成型可能なポリマー,● 可燃性・爆発性(多くの像の命を救ったが多く ビリヤード場を騒がせた,● 酢酸セルロースへの転換 1891 Louis Chardonnet: レーヨン = ニトロセルロース ● 絹代替品(Chardonnet Silk),● 光線を発しているかのような光沢 Hermann Staudinger 1953ノーベル賞: ● ポリマーがモノマーの繋がりあった鎖であることを始めて認識した(連鎖 重合体と縮合重合体) オーダー・メイド・ポリマーの時代へ
12-15ポリマーの合成 ラジカル重合:市販の有用な物質の合成
脈菅ガン(肝臓)の発生の原因 様々な繊維へ
スーパー接着剤: アニオン重合 電子吸引性基 電子吸引性基 共鳴安定化
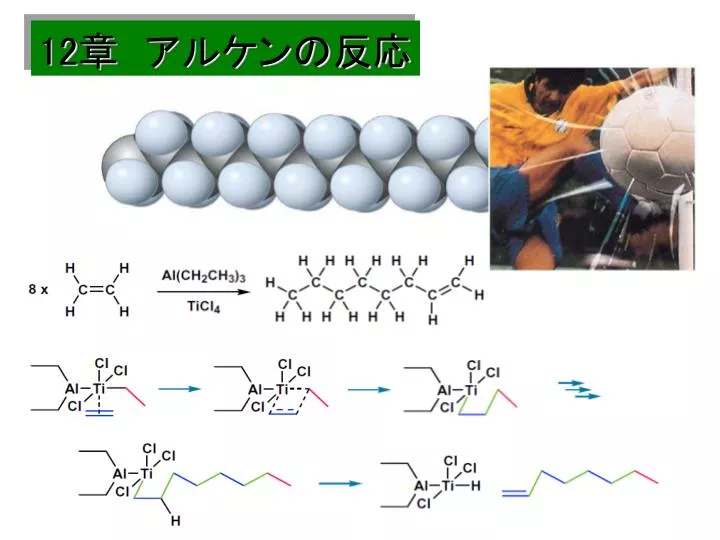
金属触媒による立体規則性重合:Ziegler-Natta による快挙(1963: Nobel Prize) 触媒 挿入 π配位 β脱離
金属触媒による重合の特徴 (1)立体規則性重合 (2)主鎖に高度な直線性 高密度ポリエチレン:耐化学薬品性,鋳型成型品 ラジカル重合の特徴(枝分かれ) 低密度ポリエチレン:しなやかで透明性大