Download

1 / 18
180 likes | 328 Views
Critical Path Initiatives. Patricia Keegan, M.D. Center for Drug Evaluation and Research FDA . Pitfalls in Drug Development. Drug Development: changing landscape Critical Path Initiatives FDA analysis of drug development programs. Approach to Review of Drug Development Programs.

E N D
Critical Path Initiatives Patricia Keegan, M.D. Center for Drug Evaluation and Research FDA
Pitfalls in Drug Development • Drug Development: changing landscape • Critical Path Initiatives • FDA analysis of drug development programs
Approach to Review of Drug Development Programs • The way FDA reviews drugs is being affected by changes in the pharmaceutical industry and advances in biology • New paradigms needed as the landscape changes
20th century Empiric discovery Nonspecific target MTD; efficacy endpoints Oncology patients will tolerate significant toxicity Patient receives information from doctor/pharmacist 21st century Designer drugs Molecular targets OBD; ? safety endpoints Trials with primary endpoints of prevention, equivalency, or QOL Direct-to-consumer advertising Approach to Drug Development Review: Changing Landscape
20th century Safety profile determined prior to approval Safety profile = tally of toxicity by body system Label = telephone directory FDA is responsible for ensuring safety of product Adverse Events U.S. vs foreign data 21st century Safety monitoring is a continuum Mechanism, PK/PD, pharmacogenetics, subgroups Better communication Risk management Ensuring the safety of a product is a shared responsibility Medication Errors International Harmonization Approach to Drug Development Review: Changing Landscape
Approach to Drug Development: Clinical Review • Determine whether the study may proceed or should be placed on hold (a legal obligation for FDA) • Provide guidance on drug development early and throughout development (a “critical path” approach that is a goal but not a requirement for FDA)
Critical Path Initiatives • Undertaken to address fall-off in new drug approvals • FDA’s analysis of problem: failure of science of drug development to keep up with science of drug discovery • Recognizes concurrent evaluation of safety, efficacy, and manufacture of new drugs
Figure 2: 10-Year Trends in Major Drug and Biological Product Submissions to FDA The figure shows the number of submissions of new molecular entities (NMEs)—drugs with a novel chemical structure—and the number of biologics license application (BLA) submissions to FDA over a 10-year period. Similar trends have been observed at regulatory agencies worldwide.
Figure 4: The Critical Path for Medical Product Development Figure 4 shows an idealized "critical path" that encompasses the drug, biological product, and medical device development processes. At the far left, ideas coming out of basic scientific research enter into an evaluation process (prototype design or discovery). In drug development the "discovery" process seeks to select or create a molecule with specific desired biological activities. Medical device development is generally much more iterative, so that prototypes often build on existing technologies. The critical path begins when candidate products are selected for development. They then undergo a series of successively more rigorous evaluation steps as they move from left to right along the path. A low percentage of candidates entering preclinical development survive to the market application stage.
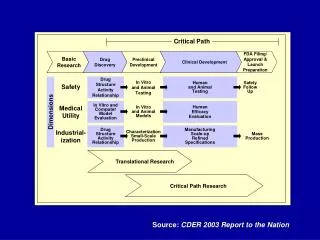
Figure 6: Working in Three Dimensions on the Critical Path Figure 6 is a highly generalized description of activities that must be successfully completed at different points and in different dimensions along the critical path. Many of these activities are highly complex—whole industries are devoted to supporting them. Not all the described activities are performed for every product, and many activities have been omitted for the sake of simplicity.
Pre-IND Considerations • Does the manufacturing process or materials raise concerns about product safety (impurities, infectious agents, lack of stability)?
Pre-IND Considerations • What is known about the product and the purported mechanism of action? • Is there evidence in the laboratory and animal studies that the product has the predicted action? If not, why not? • Does the [purported] mechanism or pathway through which drug acts raise concerns about impact on normal organs/tissues?
Initial IND Submission • Do preclinical data (or foreign experience in humans) support the safety of: • starting dose • schedule • route of administration • duration of exposure • proposed rate of dose escalation • Does the protocol appropriately monitor for toxicities (given concerns based on chemical structure, toxicologic data or prior human experience)? • Are there formulation issues, e.g., same formulation studied in animals, toxic impurities?
Phase 2 and 3 Studies • Have phase 1 studies identified an optimal dose (safety and activity) or should more dose exploration studies be done? • Are risks acceptable for population to be studied, based on non-clinical and early clinical experience?
Phase 2 and 3 Studies • Does patient selection makes sense in the context of purported mechanism of action? • Is the population easily identified or is co-development of biomarker kit needed?
Phase 2 and 3 • Do available activity data support efficacy assumptions underlying Phase 3 (registration) trials? • Are trials designed to collect efficacy data; sources of bias minimized? • Is the plan for analysis clear and appropriate?
Phase 2 and 3 Trials • Is product manufacturing sufficiently far along to support registration trials? • Are additional non-clinical studies (repro-tox or carcinogenicity) or pharmacokinetic studies (drug-drug interactions, immunogenicity, special populations) needed?
Approach to Clinical Review • Development program needs to be clear • End-of-Phase 2 meetings critical to review development to date, identify gaps, discuss future plans • Special protocol assessments permit detailed look at critical protocols within development program