Download

1 / 18
300 likes | 1.49k Views
PHM142 Fall 2012 Instructor: Dr. Jeffrey Henderson. Fumarase Deficiency. Presentation By: Claire Hooper, Paul Gillespie, Jackie Brunton , and Jacob Cashin. Focus. Introduction & History. Clinical Features & Diagnosis. Structure & Pathophysiology. Summary & Conclusion.

E N D
PHM142 Fall 2012 Instructor: Dr. Jeffrey Henderson Fumarase Deficiency Presentation By: Claire Hooper, Paul Gillespie, Jackie Brunton, and Jacob Cashin
Focus Introduction & History Clinical Features & Diagnosis Structure & Pathophysiology Summary & Conclusion
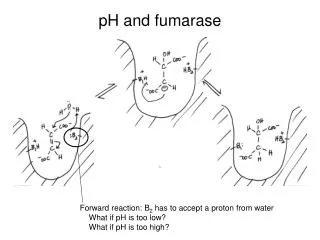
What is Fumarase? • Fumarase, also known as fumarate hydratase, is an enzyme which catalyzes the reversible hydration/dehydration of fumarate to malate • It is an important enzyme in the Krebs Cycle
Role in the Citric Acid Cycle (Krebs Cycle) - - - - Fumarate L-malate
What is Fumarase Deficiency? • Fumarase deficiency is a metabolic disorder that affects primarily the central nervous system, in particular the brain. It is a severe autosomal recessive condition and is characterized by a deficiency in the fumarase enzyme. This deficiency causes neurologic abnormalities such as severe mental retardation and autism. • It is an extremely rare disorder, with only about 100 cases worldwide (up until around 1990 there were only 13)
Treatment • Currently, there is no known cure or effective treatment for fumarase deficiency. Most people inflicted with the disease live only a few months; some live until early childhood or, in rare, cases, until early adulthood.
Historical Cases • First reported case: male infant with severe encephalopathy, delayed development • Lactic acidosis/acidemia • Urinary fumarate, succinate, and citrate levels were abnormally high • Identified attenuated levels of mitochondrial and cytosolic fumarase activity
Historical Cases • 7 month old with dementia, seizures, fumaric acidemia • Fumarate activity found to be 30-50% normal • Identified to be autosomal recessive disorder
Historical Cases • Mutation traced to FH gene • Mutations in FH also linked to renal cancer • E319Q (Glu-319Gln) • Citric acid cycle interruption • Individuals rely on anaerobic metabolism • Severe developmental consequences
Polygamist Down’s • Fumarase Deficiency has been nicknamed Polygamist Down’s • Prevalence has increased dramatically with the emergence of polygamist communities
Diagnosis • High levels of fumaric acid in the urine • 15-1000 fold higher than normal • The diagnosis is then confirmed by: • Identification of deficient fumarase activity in fibroblasts, lymphoblasts, or white blood cells • Molecular genetic testing of the FH gene • Residual enzyme activity can vary from 10-35%
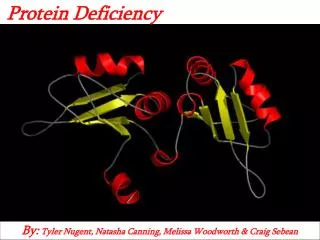
Structure of Fumarase • Homotetramer • Evolutionarily highly conserved enzyme • Central region forming alpha helix bundle – subunit interaction • Active site – enzyme function
Mutations in Fumarase Deficiency • Misense mutations most common • Frameshifts, nonsense • Deletions, insertions and duplications • Mutations within conserved regions of the enzyme • Central domain –enzyme stability • Active site – impaired enzymatic ability
Mutations in Fumarase Deficiency Picaud, S et. al. 2011 J Inherit Metab Dis, 34: 671–676.
Summary • Fumarase is an important enzyme in the citric acid cycle; converts fumarate to malate • Very rare autosomal recessive disorder resulting hindered aerobic respiratory activity; often fatal • Homotetramer • Mutations causing deficiency identified within two evolutionarily conserved region • Central alpha-helix bundle (mutations lead to defects enzyme stability) • Active site (defects in enzyme function) • Missense mutations most common • Common clinical features include global developmental delay and cerebral abnormalities and hypotonia • This disorder has been nicknamed Polygamist Down’s • This disorder is diagnosed by high levels of fumaric acid in the urine and then confirmed by either identification of the deficient fumarate hydratase enzyme and/or molecular genetic testing of the FH gene • Currently, there is no known treatment for fumarase deficiency.
References Allegri G, Fernandes MJ, Scalco FB, Correia P, et al. Fumaric aciduria: an overview and the first Brazilian case report. J Inherit Metab Dis (2010) 33:411-419 Bourgeron, T., Chretien, D., Poggi-Bach, J., Donnan, S., Rabier, D., Letouze, P., Munnich, A., Rotif, A., Landrieu, P., Rustin, P. (1994). J Clin Invest. 93(6):2514-8 Coughlin, E., Christensen, E., Kunz, P., Krishnamoorthy, K., Walker, V., Dennis, N., et al. (1998). Molecular Analysis and Prenatal Diagnosis of Human Fumarase Deficiency. Molecular Genetics and Metabolism, 63: 254–262. Deschauer, M., Gizatullina, Z., Schulze, A., Pritsch, M., Knöppel, C., Knape, M., et al. (2006). Molecular and Biochemical Investigations in Fumarase Defciency. Molecular Genetics and Metabolism, 88: 146–152. Gellera, C., Uziel, G., Rimoldi, M., Zeviani, M., Laverda, A., Carrara, F. and DiDonato, S. (1990). Fumarase deficiency is an autosomal recessive encephalopathy affecting both the mitochondrial and cytosolic enzymes. Neurology. 40:495-9 Kerrigan JF, Aleck KA, Tarby TJ, Bird CR, Heidenreich RA. Fumaric aciduria: clinical and imaging features. Ann Neurol 2000;47:583–588 Picaud, S., Kavanagh, K., Yue, W., Lee, W., Muller-Knapp, S., Gileadi, O., et al. (2011). Structural Basis of Fumarase Hydratase Deficiency. J Inherit Metab Dis, 34: 671–676. Shih VE, Mandell R. Fumarate Hydratase Deficiency. 2006 Jul 5 [Updated 2009 Jun 2]. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993 Zinn, A. B., Kerr, D. S. and Hoppel, C. L. (1986). Fumarase deficiency: a new cause of mitochondrial encephalomyopathy. N Engl J Med. 315(8): 469-75 http://oregonstate.edu/instruct/bb451/winter08/lectures/citricacidcycleoutline.html