Download
1 / 1
10 likes | 133 Views
Motivation

E N D
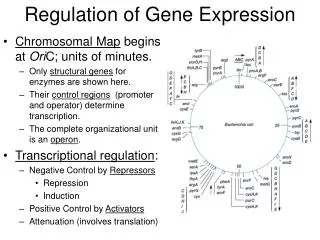
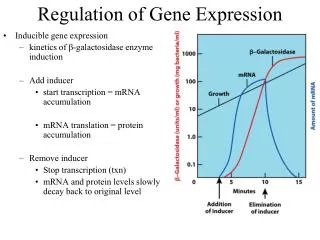
Motivation Gene regulatory networks control structure and functions of cells, it is the basis of cell differentiation, morphogenesis and adaptation. DNA microarray technology provides us with huge amount of data gene expression. There are a number of methods that use data from microarray time series for construction mathematical models of gene expression regulation. Main assumption for such methods is: if one gene (regulator gene) somehow affects the level of expression of another (target gene) then we can establish some form of relationship between their expression profiles. Main question: what kind of relationship most closely reflects the real biological process. This question has not definite answer yet. In this work we have compared linear model with time delay and nonlinear model to simulate gene expressions. Objectives: compare linear and nonlinear [1] differential models of gene expression regulation study various data processing methods influence on the models behavior test and compare the considered models on the base of some available data [2] Optimization methods: Different types of optimization algorithms were tested: simulated annealing, evolutionary algorithm and advanced method of gradient descent. Good speed and accuracy evolutionary algorithm described in [3] has shown. Comparison of models: Models were tested on the Saccharomyces cerevisiae gene expression [2], measured as amounts of mRNA using microarrays at 18 time points over two cell cycle periods (one measurement every 7 minutes for 119 minutes). We selected small subset composed of 5 target (with probes: YER1059W, YFR057W, YAL040C, YPR119W, YPL163C) and 4 regulator genes (YMR016C, YPL075W, YIL131C, YER111C). Real relationship between genes, represented by these probes were obtained from YEASTRACT open database. For four of five target genes the best regulator, found by nonlinear model, is real regulator of corresponding target, according to YEASTRACT, but only two best regulators found by linear model confirmed as real. Linear model with time delay Comparison of methods for reconstruction of models for gene expression regulation z(t) — target gene expression y(t) — regulator gene expression a, b and Δ — model parameters This model differs from more commonly used linear model: dz/dt = a + b × y(t), by presence of parameter Δ, which is introduced to reflect delay in effect exerted on target by regulator gene. Nonlinear model z(t) — target gene expression y(t) — regulator gene expression k1 — parameter reflecting the maximum level of expression k2 — parameter expressing the intensity of degradation w — weight of the regulator b — parameter responsible for transcription initiation delay, caused by independent from the regulator gene effects A.A. Shadrin1,*, I.N. Kiselev,1F.A. Kolpakov2,1 1Technological Institute of Digital Techniques SB RAS, Novosibirsk, Russia 2Institute of Systems Biology, Novosibirsk, Russia *Corresponding author: inter.cm@mail.ru Data smoothing methods Implementation of both models requires representation of gene profiles in continuous function form. The nature of microarray data (processing and measurement errors) makes interpolation inefficient. Thus to obtain gene profile continuous representation and to suppress data noise, smoothing methods are preferable. Tested smoothing methods: smoothing cubic spline with given maximum deviation in the node ("corridor spline") smoothing cubic spline with fixed weight parameter nuclear smoothing with Epanechnikov core least squares smoothing method with a basis of Chebyshev polynomials shows YPR119W target profile, reconstructed bylinear (above) and nonlinear (below) models. Each figure contains initial YPR119W profile from microarray, and its best and worst reconstruction. • Results: • The study performed on the example of two models demonstrated best results when spline with a fixed weighting parameter and the evolutionary algorithm [3] were used. • The major virtue of linear model is it’s computational simplicity and opportunity to process large amount of data. • Nonlinear model is much more harder computationally, but it allows to obtain results of higher quality by taking into account biological specificity of the process. • The models were implemented within BioUML workbench, freely available on website: http://www.biouml.org. • Conclusion: • Choice of methods for data smoothing and parameters optimization could strongly influence the behavior of the model under other conditions, hence, each study requires individual approach to select the most (or more) optimal methods. • Information, obtained with any model along, has very limited value, to get reliable information about gene interactions many versatile sources should be involved. Acknowledgments: This work was supported by EU grants FP6 №037590 “Net2Drug” and FP7 №202272 “LipidomicNet”. References: TraThi Vu, JiriVohradsky (2007) Nonlinear differential equation model for quantification of transcriptional regulation applied to microarray data of Saccharomycescerevisiae. Nucleic Acids Research, Vol. 35, No. 1. Spellman,P.T., Sherlock,G., Zhang,M., Iyer,V.,Anders,K., Eisen,M., Brown,P., Botstein,D. and Futcher,B. (1998) Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomycescerevisiae by microarray hybridization. Mol. Biol. Cell, 9, 3273–3297. Thomas P. Runarsson, Xin Yao. Stochastic Ranking for Constrained Evolutionary Optimization. IEEE Transactions on evolutionary computation, vol. 4, No. 3, september 2000. These methods are demonstrated on Above: smoothing of YIL131C probe profile from microarray, used in [2]. Below: derivative of smoothing.