Download
1 / 51
530 likes | 969 Views
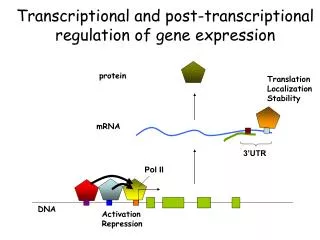
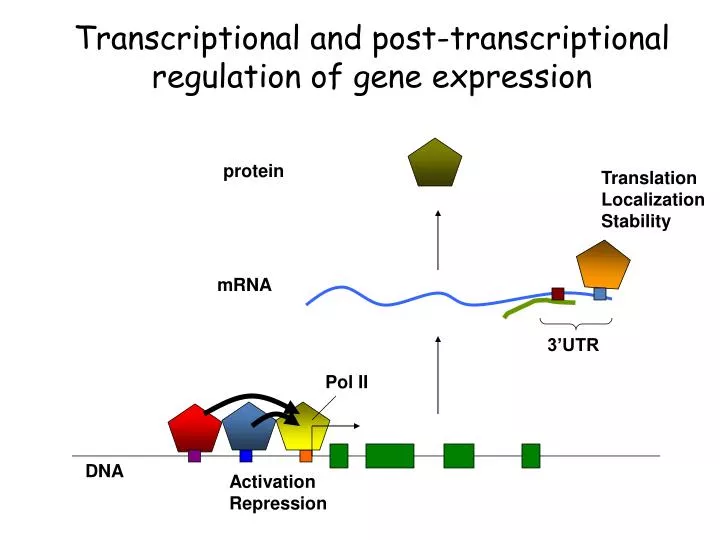
Transcriptional and post-transcriptional regulation of gene expression. protein. Translation Localization Stability. mRNA. 3’UTR. Pol II. DNA. Activation Repression. Where does each transcription factor bind in the genome, in each cell type, at a given time ? Near which genes ?

E N D
Transcriptional and post-transcriptional regulation of gene expression protein Translation Localization Stability mRNA 3’UTR Pol II DNA Activation Repression
Where does each transcription factor bind in the genome, in each cell type, at a given time ? Near which genes ? • What is the cis-regulatory code of each factor ? Does they require any co-factors ? DNA Activation Repression
ChIP-seq Transcription factor of interest Antibody Genome Analyzer II (Solexa)
Control: input DNA Genome Analyzer II (Solexa)
ACCAATAACCGAGGCTCATGCTAAGGCGTTAGCCACAGATGGAAGTCCGACGGCTTGATCCAGAATGGTGTGTGGATTGCCTTGGAACTGATTAGTGAATTCACCAATAACCGAGGCTCATGCTAAGGCGTTAGCCACAGATGGAAGTCCGACGGCTTGATCCAGAATGGTGTGTGGATTGCCTTGGAACTGATTAGTGAATTC TGGTTATTGGCTCCGAGTACGATTCCGCAATCGGTGTCTACCTTCAGGCTGCCGAACTAGGTCTTACCACACACCTAACGGAACCTTGACTAATCACTTAAG Average length ~ 250bp
25-40bp ACCAATAACCGAGGCTCATGCTAAGGCGTTAGCCACAGATGGAAGTCCGACGGCTTGATCCAGAATGGTGTGTGGATTGCCTTGGAACTGATTAGTGAATTC TGGTTATTGGCTCCGAGTACGATTCCGCAATCGGTGTCTACCTTCAGGCTGCCGAACTAGGTCTTACCACACACCTAACGGAACCTTGACTAATCACTTAAG Average length ~ 250bp
25-40bp ACCAATAACCGAGGCTCATGCTAAGGCGTTAGCCACAGATGGAAGTCCGACGGCTTGATCCAGAATGGTGTGTGGATTGCCTTGGAACTGATTAGTGAATTC TGGTTATTGGCTCCGAGTACGATTCCGCAATCGGTGTCTACCTTCAGGCTGCCGAACTAGGTCTTACCACACACCTAACGGAACCTTGACTAATCACTTAAG Average length ~ 250bp
BCL6 ChIP-seq • Lymphoma cell line (OCI-Ly1) • Solexa/Illumina • 6 lanes for ChIP, 1 for input DNA, 1 for QC • 36nt long sequences • 32 Million reads • Aligned/mapped to hg18 with Eland Melnick lab at WCMC
Read mapping with Eland Solexa Read AAAATACGCGTATTCTCCCAAAACAATATC AAAAATTCTCCCAAAACAAAAAAATACGCGTATTCTCCCAAAACAATATCTTACAAGATGTAAATATACCCAAGATG Reference Human Genome (hg18)
Read mapping with Eland Solexa Read AAAATACGCCTATTCTCCCAAAACAATATC AAAAATTCTCCCAAAACAAAAAAATACGCGTATTCTCCCAAAACAATATCTTACAAGATGTAAATATACCCAAGATG Reference Human Genome (hg18)
Read mapping with Eland Solexa Read AAAATACGCCTATTCTCCCATAACAATATC AAAAATTCTCCCAAAACAAAAAAATACGCGTATTCTCCCAAAACAATATCTTACAAGATGTAAATATACCCAAGATG Reference Human Genome (hg18)
Reads can map to multiple locations/chromosomes Solexa Read 2 Solexa Read 1 Reference Human Genome (hg18)
Reads map to one strand or the other Solexa Read 2 Solexa Read 1 hg18
>HWI-EAS83_30UCEAAXX:1:2:915:1011 AGGTCACAAAACAAGTCCTAACAAATTTAAGAGTAT U0 1 13 62 chr8.fa 59699745 R DD>HWI-EAS83_30UCEAAXX:1:2:915:1011 AGGTCACAAAACAAGTCCTAACAAATTTAAGAGTAT U0 1 13 62 chr8.fa 59699745 R DD >HWI-EAS83_30UCEAAXX:1:2:826:1245 GTCAGAAAAATCCTTTTTATTATATAAACAATACAT U2 0 0 1 chr5.fa 121195098 F DD 15G 20G >HWI-EAS83_30UCEAAXX:1:2:900:945 GTCATCAAACTCCAAGGATTCTGTTTTCAACATACT U0 1 1 0 chr18.fa 8914049 R DD >HWI-EAS83_30UCEAAXX:1:2:1037:1118 GAAAGTGATTAGCAGATTGTCATTTAATAATTGTCT U2 0 0 1 chr1.fa 97496963 F DD 18G 28G >HWI-EAS83_30UCEAAXX:1:2:898:874 GATAAATTTTTTCCTACAATCTTAAATTATTACACA U1 0 1 0 chr3.fa 95643444 R DD 10C >HWI-EAS83_30UCEAAXX:1:2:918:928 AAAAATTAAACAATTCTAAAAATATTTTTATCTTAA U2 0 0 1 chr2.fa 177727639 R DD 18C 31G >HWI-EAS83_30UCEAAXX:1:2:1324:4 GCACATGTCATACTCTTTCTAGCTCTCTTATTTTTC U0 1 0 0 chr8.fa 79132719 R DD >HWI-EAS83_30UCEAAXX:1:2:899:1015 AAATTAATGTAAAAAATAGGATACTGAATTGTGATA U1 0 1 0 chr10.fa 69774166 F DD 30G >HWI-EAS83_30UCEAAXX:1:2:909:926 GTAGTTAACAATAATTTATTTTATACTTCAAAATTC U1 0 1 17 chrX.fa 26496842 R DD 7A >HWI-EAS83_30UCEAAXX:1:2:701:1702 GTCAGAATTAATTAATCAAAACACCAAATGTACTTC U0 1 0 0 chr12.fa 72700465 F DD >HWI-EAS83_30UCEAAXX:1:2:996:1003 ATTTTGACTTTATTATTTTTTCTTCAATGTTTTTAA NM 0 0 0 >HWI-EAS83_30UCEAAXX:1:2:884:1090 GAAAGTACATCAAATACATATTATATACTTTACATA R2 0 0 2 >HWI-EAS83_30UCEAAXX:1:2:911:937 AATCCATATACATTTCTTTTTAATCATTTCCTCTTT U1 0 1 0 chr11.fa 94204222 F DD 20G >HWI-EAS83_30UCEAAXX:1:2:1517:330 GTGAGTTTCTTAATCCTGAGTTCTAATTTTATTTCA R0 29 255 255 >HWI-EAS83_30UCEAAXX:1:2:904:1031 ACATTTTATAAATTTTTAATTTCATTTTAATTTATA NM 0 0 0 >HWI-EAS83_30UCEAAXX:1:2:1291:1469 GTTTTTAAAATCAACACTTTTATTATAGAAGTAGCA U0 1 0 1 chr12.fa 62166701 R DD >HWI-EAS83_30UCEAAXX:1:2:1697:828 GTACTGATGTAAACTTGGTAAAAACATTGACATAAA U0 1 0 0 chr14.fa 65160857 F DD >HWI-EAS83_30UCEAAXX:1:2:1415:583 GAAGAAAATGACTATGTCAAAATATTATCTCTCAAT U0 1 0 0 chr5.fa 97782464 F DD >HWI-EAS83_30UCEAAXX:1:2:1561:1653 GTTTTACTGATTTTCTTACTTACTAAACTACCTGTT U0 1 0 0 chr7.fa 133200265 F DD >HWI-EAS83_30UCEAAXX:1:2:1579:943 AATGATACGGCGACCACCGACAGGTTCAGAGTTCTA NM 0 0 0 >HWI-EAS83_30UCEAAXX:1:2:1705:268 GAGAATTATTCAGAAGTCAAATCTGTGCTTAGTTTA U2 0 0 1 chr5.fa 162472124 R DD 3G 7C >HWI-EAS83_30UCEAAXX:1:2:1489:318 GTATGTATCATATATATTTATGTATCATATATATTT R1 0 3 2 >HWI-EAS83_30UCEAAXX:1:2:1003:1113 GATTGCTCCATTATTTGTTAAAAACATAGTAAAATA NM 0 0 0 >HWI-EAS83_30UCEAAXX:1:2:895:1072 ATGAGATCAGTACTTCAAAGAGATATCTGCACTCCC U0 1 1 9 chr12.fa 33830898 R DD >HWI-EAS83_30UCEAAXX:1:2:853:1178 GTTAGTCCCAATATTCCATTAATCCCAATAAATATA U2 0 0 1 chr6.fa 110722427 F DD 15G 19G >HWI-EAS83_30UCEAAXX:1:2:1432:972 GAGATAATAATAGCAGTTATGGCATCGAGATAATTT U0 1 0 0 chr2.fa 47305609 R DD >HWI-EAS83_30UCEAAXX:1:2:1718:341 GTAGAGGGCACACATCACAAACAAGTTTCTGAGAAT R2 0 0 3 >HWI-EAS83_30UCEAAXX:1:2:1171:302 GAATATCCACTTGCAGACTTTACAAACAAATTTTTT R2 0 0 4 >HWI-EAS83_30UCEAAXX:1:2:1055:1126 GGCAGATGAAACTTCTATACACTATATTTTAGCCAG U0 1 0 0 chr13.fa 90021137 F DD >HWI-EAS83_30UCEAAXX:1:2:971:1371 GAAAGAAAAACTATTGAAAAAATAGTTACTTTCCAA U0 1 0 0 chr1.fa 74303257 R DD >HWI-EAS83_30UCEAAXX:1:2:1774:614 GTGTAGATGATATCGAGGGCATTAGAAGTAAATAGC U0 1 0 0 chr5.fa 16031200 F DD >HWI-EAS83_30UCEAAXX:1:2:1207:808 GAGAGGAAATAATAAAGATAAAAGTAGAAAAAGTGA U0 1 0 0 chr1.fa 187326417 F DD >HWI-EAS83_30UCEAAXX:1:2:1680:815 GATAATTATGTTGTTGTAATTATTGTTTGTTTTTTT U0 1 0 0 chr15.fa 46739015 R DD >HWI-EAS83_30UCEAAXX:1:2:1688:260 GTTGACAATCCAGCTGTCATAGAAACTGACTATTTT U0 1 0 0 chr12.fa 38910133 R DD >HWI-EAS83_30UCEAAXX:1:2:1051:916 AAAAATTCTCCCAAAACAACAAGATGTAAATATACC U0 1 0 0 chr3.fa 101625712 R DD >HWI-EAS83_30UCEAAXX:1:2:1771:308 GTTCTTACACTGATATGAAGAAATACCTGAGACTGG U0 1 2 67 chr2.fa 214128537 R DD >HWI-EAS83_30UCEAAXX:1:2:911:917 GAGAAACACACATATTTTTGTAAGTGCCATCACATC U1 0 1 0 chr7.fa 13668652 R DD 18C >HWI-EAS83_30UCEAAXX:1:2:1105:348 GTATTATCTAACACACAAGATGATGTTTGTTTTTAT NM 0 0 0 >HWI-EAS83_30UCEAAXX:1:2:1048:857 GAGTGTAGAAAATTTTCTGCCCTAAAATATTTGTTA U1 0 1 0 chr6.fa 74625385 F DD 13G >HWI-EAS83_30UCEAAXX:1:2:743:1729 GTATCCTAAAGTGTATCTTATGTTTTTTCATCTTCT U1 0 1 0 chr12.fa 7400023 R DD 9C >HWI-EAS83_30UCEAAXX:1:2:1287:64 AATAAAACAAATTCCAATGGCTTAGATTCTACTTAA U2 0 0 1 chr10.fa 98020799 R DD 15C 20C >HWI-EAS83_30UCEAAXX:1:2:940:1059 AAATGGTCATACTTCCCAAAGCGATCTACAGATTCA U1 0 1 29 chr3.fa 50834510 R DD 19C >HWI-EAS83_30UCEAAXX:1:2:898:1061 ACATTTCCACATTTCTGTGGAAGCCTCACAATCATT R2 0 0 2 >HWI-EAS83_30UCEAAXX:1:2:913:932 ATTAATCAACAGCAACATTAATCAACTGAATCAACA U0 1 0 0 chr2.fa 46078825 R DD >HWI-EAS83_30UCEAAXX:1:2:43:1647 GAATAAATAATCAAAACATATAATACATTTTTTTAT U1 0 1 0 chr5.fa 41496935 F DD 32G >HWI-EAS83_30UCEAAXX:1:2:1412:731 ATATACACATATATATACATATATATATACACATAT R0 47 255 255 >HWI-EAS83_30UCEAAXX:1:2:1389:1196 GAGAAGGAAATGTGTTTTCTAAGTTTCTTTATCTTC U1 0 1 0 chr4.fa 188020201 F DD 32G >HWI-EAS83_30UCEAAXX:1:2:1264:1479 GTGTAGGAAAGAAAAAAGGAGGTTGTGTAGAAAAGA U0 1 0 0 chr2.fa 192227804 F DD >HWI-EAS83_30UCEAAXX:1:2:38:890 TTTATTTAAATCTTTTAAAAANTTTTTTCCAACAAA NM 0 0 0 >HWI-EAS83_30UCEAAXX:1:2:1341:1065 GATACATATACACAAAGTAAAACTATTCAGCCTCTA U0 1 0 0 chr17.fa 51416321 F DD >HWI-EAS83_30UCEAAXX:1:2:1132:929 GAGTTGTATTAATCTTAAATTGATAATTTACCATAT U1 0 1 0 chr10.fa 2376138 F DD 24G >HWI-EAS83_30UCEAAXX:1:2:1758:275 GCATTTTAACAAAATCACCATATCTGGGTAACCATT U1 0 1 0 chr21.fa 27648337 R DD 18C >HWI-EAS83_30UCEAAXX:1:2:914:1000 GAAAGCACTTTATAATAAAACAACATTGGAGCACCT U1 0 1 0 chr8.fa 67496303 F DD 16G
Number of reads per Eland type U0 21019702 65% U1 3280059 10% U2 1007173 3% R0 3661054 11% R1 815275 2% R2 406002 1% NM 20504996% QC 306352 1%
Peak detection • Calculate read count at each position (bp) in genome • Determine if read count is greater than expected
Peak detection • We need to correct for input DNA reads (control) - non-uniformaly distributed (form peaks too) - vastly different numbers of reads between ChIP and input
genome Read count Expected read count T A T T A A T T A T C C C C A T A T A T G A T A T genome Expected read count = total number of reads * extended fragment length / chr length
Is the observed read count at a given genomic position greater than expected ? Frequency x = observed read count λ = expected read count Read count The Poisson distribution
Is the observed read count at a given genomic position greater than expected ? x = 10 reads (observed) λ = 0.5 reads (expected) genome P(X>=10) = 1.7 x 10-10 log10 P(X>=10) = -9.77 -log10 P(X>=10) = 9.77 The Poisson distribution
Read count Expected read count -Log(p) Expected read count = total number of reads * extended frag len / chr len
Read count Expected read count -Log(p) Expected read count = total number of reads * extended frag len / chr len Input reads
ChIP INPUT Read count Read count Expected read count Expected read count Genome positions (bp) Genome positions (bp) -Log(Pc) -Log(Pi) Threshold Log(Pc) - Log(Pi)
Normalized Peak score (at each bp) P(XChIP) R = -log10 P(Xinput) Will detect peaks with high read counts in ChIP, low in Input Works when no input DNA !
Non-mappable fraction of the genome • chr18 9369067/76117153 0.123087459668913 (=12%) • chr2 33849240/242951149 0.139325292921335 • chr3 27854877/199501827 0.139622164963933 • chr4 27090014/191273063 0.141630052737745 • chr6 24330283/170899992 0.142365618132972 • chr8 20932821/146274826 0.143106107677065 • chr5 26029902/180857866 0.143924633059643 • chr12 19382853/132349534 0.14645199279659 • chr11 20039443/134452384 0.149044906485258 • chr20 10017788/62435964 0.160449000194824 • chr7 26182588/158821424 0.164855517225434 • chr10 22968951/135374737 0.169669404417753 • chr17 14496284/78774742 0.184021980040252 • chrX 31269270/154913754 0.201849540099583 • chr1 55186693/247249719 0.223202247602959 • chr13 28668063/114142980 0.251159230291692 • chr16 23552340/88827254 0.265147676410215 • chr14 29689825/106368585 0.279122120502026 • chrM 4628/16571 0.279283084907368 • chr9 43125838/140273252 0.307441635415995 • chr19 20251255/63811651 0.317359834491667 • chr15 31877970/100338915 0.317702957023205 • chr21 16867677/46944323 0.359312392256674 • chr22 21176578/49691432 0.426161556382597 • chrY 43209644/57772954 0.747921665906161 (=74%) We enumerated all 30-mers, counted # occurrences, calculated non-unique fraction of genome
Peak detection • Determine all genomic regions with R>=15 • Merge peaks separated by less than 100bp • Output all peaks with length >= 100bp • Process 23M reads in <7mins
BCL6: 18,814 peaks ChIP reads Input reads Detected Peaks 80% are within <20kb of a known gene
Where does each transcription factor bind in the genome, in each cell type, at a given time ? Near which genes ? • What is the cis-regulatory code of each factor ? Does they require any co-factors ? DNA Activation Repression
Discovering regulatory sequences associated with peak regions True TF binding peak? Yes correlation is quantified using the mutual information Yes Target regions Yes True TF peak Yes Yes No Yes Yes … Absent Motif No Present No Random regions No No No No …
Highly informative k-mer MI CTCATCG 0.0618 TCATCGC 0.0485 AAAATTT 0.0438 GATGAGC 0.0434 AAAAATT 0.0383 ATGAGCT 0.0334 TTGCCAC 0.0322 TGCCACC 0.0298 ATCTCAT 0.0265 ... ... ACGCGCG 0.0018 CGACGCG 0.0012 TACGCTA 0.0011 ACCCCCT 0.0010 CCACGGC 0.0009 TTCAAAA 0.0005 AGACGCG 0.0004 CGAGAGC 0.0003 CTTATTA 0.0002 MI=0.081 MI=0.045 MI=0.040 ... Not informative Motif Search Algorithm
A/G C/G/T T/G A/T/G C/G A/C/G Optimizing k-mers into more informative degenerate motifs True TF binding peak? ATCCGTACA Yes Yes Target regions Yes Yes Yes Yes … No No Random regions ATCC[C/G]TACA No No No which character increases the mutual information by the largest amount ? No …
A/C C/G/T T/C A/T/C C/G A/C/G Optimizing k-mers into more informative degenerate motifs True TF binding peak? Yes Yes Target regions Yes Yes Yes Yes … No No ATCC[C/G]TACA Random regions No No . . . No No …
Mutual information change Similarity to ChIP-chip RAP1 motif Motif Conservation with S. bayanus
Motifs optimized so far k-mer MI CTCATCG 0.0618 TCATCGC 0.0485 AAAATTT 0.0438 GCTCATC 0.0434 AAAAATT 0.0383 ATGAGCT 0.0334 TTGCCAC 0.0322 TGCCACC 0.0298 ATCTCAT 0.0265 ... MI=0.081 Highly informative k-mers MI=0.045 optimize ? Only optimize k-mer if I(k-mer;expression | motif) is large enough (for all motifs optimized so far) Conditional mutual information I(X;Y|Z)
Motif co-occurrence anallysis Discovered Motifs Enrichment Depletion FIRE automatically compares discovered motifs to known motifs in TRANSFAC and JASPAR
ChIPseeqer: an integrated framework for ChIP-seq data analysis • ChIPseeqer (peak detection) • ChIPseeqer2Track (for Genome Browser) • ChIPseeqer2FIRE (+ motif analysis) • ChIPseeqer2iPAGE (+ pathway analysis) • ChIPseeqer2cons (conservation analysis)
Installing and setting up programs Install ChIPseeqer and FIRE: http://physiology.med.cornell.edu/faculty/elemento/lab/chipseq.shtml http://tavazoielab.princeton.edu/FIRE/ Execute following commands: export FIREDIR=/Applications/FIRE-1.1 export PATH=$PATH:$FIREDIR export CHIPSEEQERDIR=/Applications/ChIPseeqer-1.0 export PATH=$PATH:$CHIPSEEQERDIR:$CHIPSEEQERDIR/SCRIPTS chmod +x $CHIPSEEQERDIR/ChIP* chmod +x $CHIPSEEQERDIR/SCRIPTS/*.pl
Peak Detection - Input file: CTCF.bed cd ~/Desktop/elemento Or download from: http://physiology.med.cornell.edu/faculty/elemento/lab/files/chipseq/ - 2947043 U0 reads in BED format (check by typing wc –l CTCF.bed) (view by typing more CTCF.bed and q to exit) - No input DNA for this experiment
Peak Detection Step 1: Split big read file into one file per chromosome split_bed_or_mit_files.pl CTCF.bed Expected output: Opening CTCF.bed Current directory = . Creating ./reads.chr1 …
Peak Detection Step 2. Detect peaks ChIPseeqer --chipdir=. --t=15 --fraglen=250 --format=bed -outfile=CTCF_peaks_t15.txt Expected output: Processing reads in chrY ... done. Processing reads in chrX ... done. Processing reads in chr9 ... done. Processing reads in chr8 ... done. Step 3. Count how many peaks were found wc -l CTCF_peaks_t15.txt
Making a Genome Browser track Command lines: cd JuliaChild wc –l CTCF_peaks_t15.txt ChIPseeqer2track --targets=CTCF_peaks_t15.txt --trackname=“CTCF peaks” Expected output: CTCF_peaks_t15.txt.wgl.gz created. To check that the file was created: ls
Making a Genome Browser track http://genome.ucsc.edu/cgi-bin/hgGateway
Making FIRE input files Command line (type instructions below as one single line): ChIPseeqer2FIRE --targets=CTCF_peaks_t15.txt –genome=wg.fa --suffix=CTCF_peaks_t15_FIRE wg.fa is also available from: http://physiology.med.cornell.edu/faculty/elemento/lab/files/chipseq/ (decompress with gunzip wg.fa.gz) Expected output: Extracting sequences ... Done. Extracting randomly selected sequences ... Done. CTCF_peaks_t15_FIRE.txt and CTCF_peaks_t15_FIRE.seq have been generated. …
FIRE analysis Command line (type instructions below as one single line): fire.pl --expfile=CTCF_peaks_t15_FIRE.txt --fastafile_dna=CTCF_peaks_t15_FIRE.seq --nodups=1 --minr=2 --species=human --dorna=0 --dodnarna=0 Expected output: Extracting sequences ... Done. Extracting randomly selected sequences ... Done. CTCF_peaks_t15_FIRE.txt and CTCF_peaks_t15_FIRE.seq have been generated. …
FIRE main output file open CTCF_peaks_t15_FIRE.txt_FIRE/DNA/CTCF_peaks_t15_FIRE.txt.summary.pdf Randomly selected sequences Peak sequences




![[VI]. Post-Transcriptional Processing and Post-Transcriptional Control of Gene Expression](https://cdn1.slideserve.com/3214110/vi-post-transcriptional-processing-and-post-transcriptional-control-of-gene-expression-dt.jpg)






![[V]. Process of Transcription and Transcriptional Control of Gene Expression](https://cdn2.slideserve.com/5058527/v-process-of-transcription-and-transcriptional-control-of-gene-expression-dt.jpg)