Download
1 / 42
420 likes | 552 Views
Was kann ich per Knopfdruck über eine PDB-Struktur lernen?. PdbSum webseite: http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/. Klassifizierung in CATH. http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/. Darstellung der Sekundärstruktur.

E N D
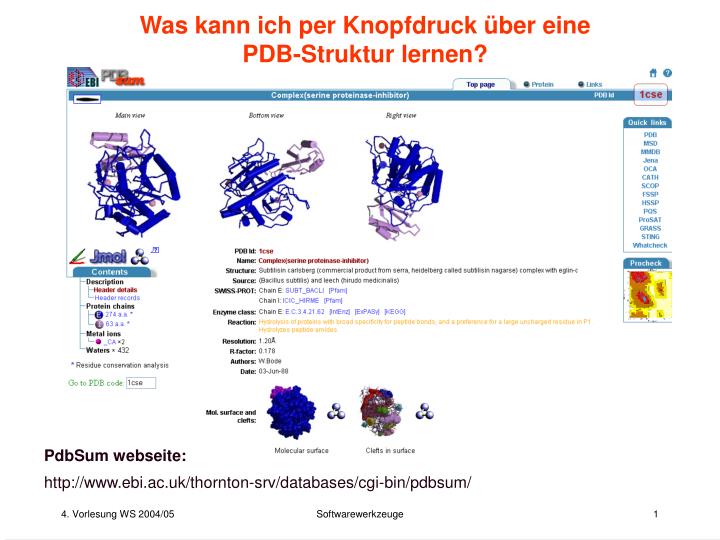
Was kann ich per Knopfdruck über eine PDB-Struktur lernen? PdbSum webseite: http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/
Klassifizierung in CATH http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/
Darstellung der Sekundärstruktur http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/
Konservierung innerhalb Proteinfamilie Oberfläche entsprechend Konservierung eingefärbt. http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/
Multiples Sequenzalignment http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/
Ramachandran-Diagramm http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/
Oberfläche Spalten (clefts) auf Oberfläche sind mögliche Bindungstaschen! http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/
Sekundärstrukturvorhersage: PSIPRED Enge, sehr polare Bindungstasche auf Proteinoberfläche. D.T. Jones, J Mol Biol 292, 195 (1999); http://bioinf.cs.ucl.ac.uk/psipred/
Qualität von PSIRED-Vorhersagen Ergebnis für 187 Testproteine mit unterschiedlichen Faltungen. Genauigkeit von PSIPRED: Ca. 75% D.T. Jones, J Mol Biol 292, 195 (1999); http://bioinf.cs.ucl.ac.uk/psipred/
Vorhersage von TM-Helices Residuen in Transmembranhelices sind fast ausschließlich hydrophob. Länge einer TM-Helix ≥ 20 Residuen. HMMs sind sehr erfolgreich um TM-Helices vorherzusagen (>90% Genauigkeit). http://darwin.nmsu.edu/~molb470/ fall2003/Projects/koul/tmhmm.html
Analyse der Oberfläche: elektrostatisches Potential Proteinoberflächen an Protein-Protein-Bindungsstellen sind häufig elektrostatisch komplementär. Surface representation of the electrostatic potential of unbound monomers of 4 protein-protein complexes. Open book view of the protein–protein interfaces is shown. Color range from deep red to deep blue corresponds to the range in the values of electrostatic potential from −10 to +10kT/e, where k is the Boltzmann constant, T is the absolute temperature and e is a proton's charge. Sheinerman, Honig, J Mol Biol 318, 161 (2002)
PROCHECK: Qualitätscheck für Proteinstrukturen The Ramachandran plot shows the phi-psi torsion angles for all residues in the structure (except those at the chain termini). Glycines are separately identified by triangles as these are not restricted to the regions of the plot appropriate to the other sidechain types. Colouring/shading scheme: the darkest areas (here shown in red) correspond to the "core" regions representing the most favourable combinations of phi-psi values. The regions are labelled as follows: A - Core alpha B - Core beta L - Core left-handed alpha p - Allowed epsilon a - Allowed alpha b - Allowed beta l - Allowed left-handed alpha ~a - Generous alpha ~p - Generous epsilon ~l - Generous left-handed alpha ~b - Generous beta The different regions were taken from the observed phi-psi distribution for 121,870 residues from 463 known X-ray protein structures. The two most favoured regions are the "core" and "allowed" regions which correspond to 10° x 10° pixels having more than 100 and 8 residues in them, respectively. The "generous" regions were defined by Morris et al. (1992) by extending out by 20° (two pixels) all round the "allowed" regions. In fact, the authors found very few residues in these "generous" regions, so they can probably be treated much like the "disallowed" region and any residues in them investigated more closely. Ideally, one would hope to have over 90% of the residues in the "core" regions. The percentage of residues in the "core" regions is one of the better guides to the stereochemical quality of a protein structure. http://www.biochem.ucl.ac.uk/~roman/procheck
PROCHECK The plot shows separate Ramachandran plots are shown for each of the 20 different amino acid types. The darker the shaded area on each plot, the more favourable the region. The data on which the shading is based has come from a data set of 163 non-homologous, high-resolution protein chains chosen from structures solved by X-ray crystallography to a resolution of 2.0Å or better and an R-factor no greater than 20%. The numbers in brackets, following each residue name, show the total number of data points on that graph. The red numbers above the data points are the reside-numbers of the residues in question (ie showing those residues lying in unfavourable regions of the plot). http://www.biochem.ucl.ac.uk/~roman/procheck
PROCHECK: analysis of side chain angles http://www.biochem.ucl.ac.uk/~roman/procheck
PROCHECK The 6 graphs show how the structure (represented by the solid square) compares with well-refined structures at a similar resolution. The dark band in each graph represents the results from the well-refined structures; the central line is a least-squares fit to the mean trend as a function of resolution, while the width of the band on either side of it corresponds to a variation of one standard deviation about the mean. In some cases, the trend is dependent on the resolution, and in other cases it is not. The 6 properties plotted are: a. Ramachandran plot quality. This property is measured by the percentage of the protein's residues that are in the most favoured, or core, regions of the Ramachandran plot. For a good model structure, obtained at high resolution, one would expect this percentage to be over 90%. However, as the resolution gets poorer, so this figure decreases - as might be expected. The shaded region reflects this expected decrease with worsening resolution. b. Peptide bond planarity. This property is measured by calculating the standard deviation of the protein structure's omega torsion angles. The smaller the value the tighter the clustering around the ideal of 180 degrees (which represents a perfectly planar peptide bond). c. Bad non-bonded interactions. This property is measured by the number of bad contacts per 100 residues. Bad contacts are selected from the list of non-bonded interactions and are defined as contacts where the distance of closest approach is less than or equal to 2.6Å. d. Calpha tetrahedral distortion. This property is measured by calculating the standard deviation of the zeta torsion angle. This is a notional torsion angle in that it is not defined about any actual bond in the structure. Rather, it is defined by the following four atoms within a given residue: Calpha, N, C, and Cbeta. e. Main-chain hydrogen bond energy. This property is measured by the standard deviation of the hydrogen bond energies for main-chain hydrogen bonds. The energies are calculated using the method of Kabsch & Sander (1983). f. Overall G-factor. The overall G-factor is a measure of the overall normality of the structure. The overall value is obtained from an average of all the different G-factors for each residue in the structure. http://www.biochem.ucl.ac.uk/~roman/procheck
PROCHECK The 5 properties plotted are: a. Standard deviation of the chi-1 gauche minus torsion angles. b. Standard deviation of the chi-1 trans torsion angles. c. Standard deviation of the chi-1 gauche plus torsion angles. d. Pooled standard deviation of all chi-1 torsion angles. e. Standard deviation of the chi-2 trans torsion angles. http://www.biochem.ucl.ac.uk/~roman/procheck
PROCHECK Distributions of each of the different main-chain bond lengths in the structure. The solid line in the centre of each plot corresponds to the small-molecule mean value, while the dashed lines either side show the small-molecule standard deviation, the data coming from Engh & Huber (1991). Highlighted bars correspond to values more than 2.0 standard deviations from the mean, though the value of 2.0 can be changed by editing the procheck.prm file. http://www.biochem.ucl.ac.uk/~roman/procheck
PROCHECK Distributions of each of the different main-chain bond angles in the structure. The solid line in the centre of each plot corresponds to the small-molecule mean value, while the dashed lines either side show the small-molecule standard deviation, the data coming from Engh & Huber (1991). If any of the histogram bars lie off the graph, to the left or to the right, a large arrow indicates the number of these outliers (as in the CA-C-O and CB-CA-C plots above). http://www.biochem.ucl.ac.uk/~roman/procheck
PROCHECK RMS distances from planarity for the different planar groups in the structure. The dashed lines indicate different ideal values for aromatic rings (Phe, Tyr, Trp, His) and for planar end-groups (Arg, Asn, Asp, Gln, Glu). The default values are 0.03Å and 0.02Å, respectively. http://www.biochem.ucl.ac.uk/~roman/procheck
Wie kann man 2 Proteinstrukturen vergleichen? Paarweise Sequenzvergleiche Paarweise Strukturvergleiche?
Partitioning protein space into homologous families Protein architecture. The tramtrack protein [Protein Data Bank entry 2drp (30)] is a small protein (525 heavy atoms, 63 residues, and 6 elements of secondary structure), yet it exhibits typical modular protein architecture with two compact structural domains, the so-called zinc fingers. (A) The most detailed description of atomic positions is required to understand the function of the tramtrack protein (gray and black, running left to right), which involves binding to a specific base sequence of DNA (white). Holm, Sander Science 273, 5275 (1996)
Partitioning protein space into homologous families (B) The complicated 3D shape of proteins is encoded in their linear sequence of amino acids. Side chains stripped off, the polypeptide backbone (thick) can be seen meandering from the bottom left to the upper right. Regular patterns of hydrogen bonding (thin lines) between amide and carbonyl groups of the polypeptide backbone give rise to secondary structure, shown schematically in (C) as arrows for strands and cylinders for helices (with zinc atoms as spheres). Holm, Sander Science 273, 5275 (1996)
Meaning of structural equivalence Shape comparison aims at the 1:1 enumeration of equivalent polymer units in 2 protein molecules. The problem and solution can be represented in 3D, as a rigid-body superimposition; in 2D, as similar patterns in distance matrices; or in 1D, as an alignment of amino acid sequences. Here, the comparison of the tramtrack protein with another zinc finger protein, the human enhancer-binding protein MBP-1 [PDB entry 1bbo], is used as an example. (A) In the 3D comparison, the problem is to find a translation and rotation of one molecule (red: 1bbo) onto the other (blue: 2drpA). The 3D superimposition (residue centers only, green lines join equivalenced residue centers, zinc atoms as spheres) is not exact because of an internal rotation of the two zinc finger domains relative to one another. Holm, Sander Science 273, 5275 (1996)
Ranges of similarity between proteins Holm et al. Prot Sci 1, 1691 (1992)
Surprising similarities Holm et al. Prot Sci 1, 1691 (1992)
Surprising similarities Holm et al. Prot Sci 1, 1691 (1992)
Surprising similarities Holm et al. Prot Sci 1, 1691 (1992)
Partitioning protein space into homologous families (B) The 2D distance matrices reveal the conserved structure of the zinc fingers (left: distance matrices of the whole structures; black dots are intramolecular distances less than 12 Å, 1bbo at bottom and 2drpA on top; right: distance matrices brought into register by keeping only rows or columns corresponding to structurally equivalent residues). (C) One-dimensional alignment of amino acid strings. Evolutionary comparison aligns the histidine (H) residues involved in zinc binding (bold; helices and strands of secondary structure are underlined). Holm, Sander Science 273, 5275 (1996)
2 Algorithms for structural alignment (A) The 3D lookup is a fast heuristic algorithm that catches easy-to-find structural similarities and is part of the Dali 3D search server. The idea is that in favorable cases, 3D superimposition of only a pair of secondary structure elements (SSEs) leads to superimposition of the entire structures. Top: Structure comparison of an SH3 domain of c-Src kinase [1cskA, query structure] with the enzyme papain [1ppn, target structure] reveals similar domain folds, although there is no sequence relation between the proteins and one is much larger. The appropriate orientation of the molecules is found by exhaustive comparison of internal coordinate frames of each protein. An internal coordinate frame is defined by an ordered pair of SSEs (centering one SSE at the origin, aligning it with the y axis, and rotating the molecule around this axis so that the center of a second SSE is in the positive x-y plane). Bottom left: Target structure, papain, loaded onto the SSE lookup grid. Each pair of SSEs where the segment midpoints are within 12 Å defines a coordinate frame relative to the grid axes. The figure shows the transformed positions of the 12 SSEs of papain (dotted lines) in each of the 100 different coordinate frames defined by different pairs of SSEs. Bottom right: The target lookup grid is probed with the SH3 domain, which has four SSEs (thick continuous lines). The coordinate frames shown are the ones yielding the best 3D match of four segments. Iterative extension of a residue-wise alignment starting from the preorientation defined by the SSE match shown here leads to the equivalence of 43 C atoms with 1.7 Å root-mean-square positional deviation on an optimal least-squares superimposition. Holm, Sander Science 273, 5275 (1996)
Branch-and-bound algorithm (B) A branch-and-bound algorithm is guaranteed to yield the global optimum but may, in the worst case, need an exponential number of steps to do so. An implementation of this algorithm is an essential part of the Dali 3D search server. First, protein structures A and B are represented by distance matrices (bottom left and right; each point in a matrix is a residue-residue distance; an internal square is a set of contacts made by two segments; the secondary structure segments are ,, and ). The problem of shape comparison becomes one of finding a best subset of residues in each matrix (subsets of rows and columns) such that the set of residues in protein A has a similar pattern of intramolecular distances as the set in protein A, as in Fig. 2B. A single solution to the problem is given in terms of the two sets of equivalent residues (an alignment), as shown in Fig. 2C. The solution space consists of all possible placements of residues in protein B relative to the segments of residues of protein A. The key algorithmic idea is to recursively split the solution subspace (schematically shown as a circle at upper left, in which each point is a solution to the problem and the lines divide subsets of solutions) that yields the highest upper bound until there is a single alignment trace left: start with the entire circle; calculate the upper bound for the left (9) and right (17) half; choose the right half and split it into top (upper bound 10) and bottom (upper bound 16) quarters; choose the bottom part and split it (left: 14; right: 12); choose the right part; and so on until the area of solution space has shrunk to a single solution (shown as the residue-residue alignment matrix enlarged at right). The upper bound for each part of the solution space is estimated in terms of a simplified subproblem that asks for the best match of residues in protein B onto a predefined set of residues in protein A (the match is illustrated by the circle-ended line connecting the single square in matrix A with a set of candidate squares in matrix B). The best match is the one with the maximal pair score (sum of similarities of distances between the square in A and the square in B). The predefined set corresponds to residues in secondary structure elements ( , ). The upper bound for each of the segment-segment submatrices of matrix A is found by calculating the similarity scores between the submatrix in A and all accessible submatrices in B. An upper bound of the total similarity score (sum over all segment-segment submatrices in A) for one set of solutions is given by the sum of separately calculated upper bounds for each segment-segment pair of matrix A. The method for choosing constraints that define a set of solutions works in terms of defining allowed residue ranges at each stage of the iteration and is not illustrated. Holm, Sander Science 273, 5275 (1996)
Recurrent folds (A) A small number of frequently occurring domains (folds) covers a large fraction of all known protein structures. The 287 structurally unique protein domains (folds) are ranked in descending order of occurrence in the representative set of 740 proteins. Domains ranked 1 through 16 occur 10 or more times each. Domains ranked 1 through 26 cover 50% of all known structures that is, the essential parts of these structures can be constructed from these domains or described in terms of these domains (within the limits of similarity within a domain class). Domains ranked about 170 or higher occur only once in the current database (singlets). Holm, Sander Science 273, 5275 (1996)
Partitioning protein space into homologous families (B) Examples of frequently observed fold classes, with one class from each of the attractor regions in Fig. 5 (each attractor region contains several classes, where the term "class" is defined in the text). Color coding indicates which parts of the fold are present in more or fewer members of the class. The color changes from light blue (regions present in 100% of members of the fold class) to red (0% occupancy). The representative classes are defined as follows (attractor, class name, and number of recurrences in sequence-unique set of 740 structures): attractor I: parallel : COOH-terminal domain of succinyl-CoA synthetase chain (126); attractor II: -meander: mouse opg2 immunoglobulin heavy chain variable domain (52); attractor III: -helical: myoglobin; attractor IV: -zigzag: COOH-terminal domain of pertussis toxin; and attractor V: meander: COOH-terminal domain of phosphoglycerate dehydrogenase. Note that other fold classes in the same attractor region are not shown, but the most frequently occurring are shown in Fig. 5B. Holm, Sander Science 273, 5275 (1996)
Partitioning protein space into homologous families (C) Growth and redundancy of protein 3D structures in the Protein Data Bank. Entry: one of currently more than 4000 sets of protein coordinates in the PDB. Family: collection of proteins set as equivalent if pairwise sequence identity exceeds 25%. Fold: fold class as defined above. The number of new structure entries grows rapidly in time (note logarithmic scale). Redundancy is defined in terms of sequence similarity (sequence families) or structure similarity (fold classes). Currently, there are about 6.4 entries per sequence family and 2.4 families per fold class, for a total of 15 entries per fold. One may expect that in the near future a new fold will appear for about every 15 new entries. The curve of new folds lags behind the curve of sequence-unique families, which indicates the increasing frequency of recurrent folds in newly solved structures (although this may be the result of bias in experimental work). There is no indication that the growth in new fold classes is slowing down at present. Holm, Sander Science 273, 5275 (1996)
Partitioning protein space into homologous families (B) 40% of all known domains (protein substructures) are covered by 16 fold classes (shown as topology diagrams; , -helix segment; , -strand segment; thick bar, parallel chain connection between segments; thin bars, antiparallel connection; arc, helices crossing at roughly right angles). Although each fold class has individual features, most fold classes map to five attractor regions (peaks I through V). All folds with sheets of mainly parallel strands map to attractor I. The parallel folds contain a x unit, where the intervening segment (x) is required to reverse chain direction so that the strands are parallel. The unit has a preferred handedness determined by polymer physics and the natural twist of strands. Attractor II contains a variety of helical folds. The connectivity of elements in the folds of attractors III and IV contains meander motifs suggestive of the collapse of a long hairpin, either of strands only or of strands alternating with a helical pair, ()2. The zigzag motif of attractor V is simply a series of antiparallel hairpin connections between sequentially adjacent strands. Elementary polymer physics indicates that interactions in space between regions of the chain that are close in sequence are much more probable than those between sequence-distant regions. The zigzag motif occurs both in flat sheets and barrels, and there is considerable variation in the length of strands (about 4 residues in propeller blades, about 13 in porin barrels). Holm, Sander Science 273, 5275 (1996)
Evolutionary adaptation of enzyme function (A) Discovery of an essential structure-function feature by shape comparison. A structure database search with DNA polymerase detects kanamycin nucleotidyltransferase (rather than other known DNA or RNA polymerases) as the nearest neighbor in fold space and reveals conserved residues and structural features supporting the active site. Following up the lead provided by structure database searching with profile searches in sequence databases resulted in the identification of the same characteristics in a large superfamily of nucleotidyltransferases. The biological functions of member families range from DNA repair to regulation of biosynthetic pathways and antibiotic resistance. Holm, Sander Science 273, 5275 (1996)
Partitioning protein space into homologous families (B) Variety of substrate specificity of a common chemical reaction on an essential protein substructure is the remarkable result of biological evolution. All member enzymes of this extended family unified as a result of shape comparison catalyze a common chemical reaction, the coupling of nucleoside triphosphates (black squares and dots) to a free hydroxyl group by means of elimination of pyrophosphate [top row: DNA polymerase , DNA nucleotidyl exotransferase; middle row: polyadenylate polymerase, (2‘-5‘) oligoadenylate synthetase, kanamycin nucleotidyltransferase; bottom row: protein PII uridylyltransferase, glutamine synthetase adenylyltransferase, and streptomycin 3‘-adenylyltransferase]. Holm, Sander Science 273, 5275 (1996)
Partitioning protein space into homologous families a, All-against-all structure alignment by DALI reveals a hierarchical organization of fold space. The method is sensitive enough to recognize similarities of general folding pattern — e.g., the -sandwich topology of superoxide dismutase and immunoglobulin domains — and selective enough to give higher scores to pairs of structures with more closely superimposable C traces — e.g., any two globins score higher than any globin–phycocyanin pair. Structure similarity alone yields an operational definition of 'folds'. The thick circles denoting folds (left) are defined using a uniform radius for clusters of structural neighbors. The vertical bar (right) denotes cutting the fold dendrogram at a uniform value of structural similarity. However, the level of structural similarity, or degree of structural divergence, varies between different families, and we need other criteria to delineate superfamilies. b, Divergent evolution from a common ancestor retains not only the fold but also many functional features. This means that homologs remain in a structural neighborhood and can be delineated by similar functional attributes (marked here by similar color) in the map of fold space. Functional convergence (from independent evolutionary origins) would appear as blotches of similar color in disconnected regions of the map of fold space and in disjoint branches of the fold dendrogram. Partitioning the fold dendrogram in terms of functional similarities yields family-specific thresholds in terms of structural similarity (nodes that partition the fold dendrogram into functionally conserved superfamilies are circled on the right). This combination of structural and functional similarity measures results in an automatically generated hierarchical classification m_n at the fold (m) and superfamily (n) levels. Dietmann & Holm, Nat Struct Biol 8, 953 (2001)
Proteinstruktur-Analyse c, The principles are illustrated on a branch of the fold dendrogram consisting of aminopeptidases (1xjo and 1amp), carboxypeptidase (1aye), purine nucleoside phosphorylases (1b8oA, 1cb0A and 1ecpA), pyrrolidone carboxyl peptidase (1a2zA), peptidyl–tRNA hydrolase (2pth) and hydrogenase maturating endopeptidase (1cfzA). The functional similarity between all pairs of structures is evaluated using a neural network with output in the range 0 (analogous)-1 (homologous) — for example, (1cb0A, 1b8oA) = 0.91, (1amp, 1aye) = 0.74, (1cfzA, 2pth) = 0.59, (1xjo, 1amp) = 0.30 and (1a2zA, 2pth) = 0.13. Here, line thickness indicates the magnitude of the term (i,j) - (Eq. 1) with color-coding for positive (red) or negative (blue) values. The threshold parameter was arbitrarily set to 0.30 in this numerical example. d, The protein set is partitioned into superfamilies in the context of the fold dendrogram. Node scores s(C) are computed for each node, with = 0.30. For example, each structure is homologous to itself; therefore, leaf nodes get a score s(leaf) = 1.00 - = 0.70, whereas s(1cfzA, 2pth) = (1.00 + 1.00 + (2 0.59)) / 4 - = 1.98. The optimal partition (circled nodes) maximizes the sum of node scores over selected nodes (underlined scores). This optimal partition is stable for threshold values 0.09 < < 0.53. Dietmann & Holm, Nat Struct Biol 8, 953 (2001)
Partitioning protein space into homologous families Dietmann & Holm, Nat Struct Biol 8, 953 (2001)
Proteinstruktur-Vergleich durch Feature-Vector Input für Neuronales Netzwerk ist ein Feature-Vector. „Keyword similarity“: Vektorprodukt für Häufigkeiten von Swissprot-Keywörter innerhalb der beiden Sequenzfamilien. „Functional preference“ is pro Aminosäure definiert und wird über alle Residuen in einem 3D-Cluster von konservierten Residuen summiert. Dietmann & Holm, Nat Struct Biol 8, 953 (2001)
Funktionszuordnung per Strukturvergleich Dietmann & Holm, Nat Struct Biol 8, 953 (2001)
Zusammenfassung Viele, sehr bequeme Tools verfügbar, mit denen man schnell einen guten Überblick über bestimmte Proteinstrukturen erhalten kann. Proteinstruktur ist evolutionär wesentlich länger konserviert als Sequenz Strukturvergleiche erlauben es, wesentlich entferntere Verwandtschaften aufzudecken. Numerische Klassifizierung erlaubt (nun erstmals) eine robuste, automatische evolutionäre Klassifikation von Proteinstrukturen. Dietmann & Holm, Nat Struct Biol 8, 953 (2001)

