Download
1 / 1
10 likes | 66 Views
Discover how students at Longwood University engage in finishing sequencing and annotation research projects focusing on Drosophila species to explore evolutionary genomics. Learn the step-by-step procedures involved in genome finishing and annotation processes for comparative analysis.

E N D
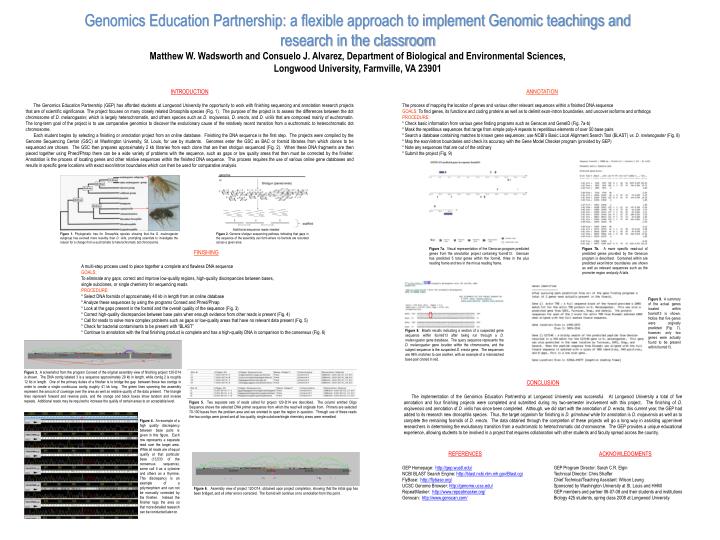
Genomics Education Partnership: a flexible approach to implement Genomic teachings and research in the classroom Matthew W. Wadsworth and Consuelo J. Alvarez, Department of Biological and Environmental Sciences, Longwood University, Farmville, VA 23901 INTRODUCTION The Genomics Education Partnership (GEP) has afforded students at Longwood University the opportunity to work with finishing sequencing and annotation research projects that are of scientific significance. The project focuses on many closely related Drosophila species (Fig. 1). The purpose of the project is to assess the differences between the dot chromosome of D. melanogaster, which is largely heterochromatin, and others species such as D. mojavensis, D. erecta, and D. virilis that are composed mainly of euchromatin. The long-term goal of the project is to use comparative genomics to discover the evolutionary cause of the relatively recent transition from a euchromatic to heterochromatic dot chromosome. Each student begins by selecting a finishing or annotation project from an online database. Finishing the DNA sequence is the first step. The projects were compiled by the Genome Sequencing Center (GSC) at Washington University, St. Louis, for use by students. Genomes enter the GSC as BAC or fosmid libraries from which clones to be sequenced are chosen. The GSC then prepares approximately 2 kb libraries from each clone that are then shotgun sequenced (Fig. 2). When these DNA fragments are then pieced together using Phred/Phrap there can be a wide variety of problems with the sequence, such as gaps or low quality areas that then must be corrected by the finisher. Annotation is the process of locating genes and other relative sequences within the finished DNA sequence. This process requires the use of various online gene databases and results in specific gene locations with exact exon/intron boundaries which can then be used for comparative analysis. ANNOTATION The process of mapping the location of genes and various other relevant sequences within a finished DNA sequence GOALS: To find genes, its functions and coding proteins as well as to delimit exon-intron boundaries, and uncover isoforms and orthologs PROCEDURE: * Check basic information from various gene finding programs such as Genscan and GeneID (Fig. 7a-b) * Mask the repetitious sequences that range from simple poly-A repeats to repetitious elements of over 50 base pairs * Search a database containing matches to known gene sequences; use NCBI’s Basic Local Alignment Search Tool (BLAST) vs.D. melanogaster (Fig. 8) * Map the exon/intron boundaries and check its accuracy with the Gene Model Checker program (provided by GEP) * Note any sequences that are out of the ordinary * Submit the project (Fig. 9) Figure 1. Phylogenetic tree for Drosophila species showing that the D. melanogaster subgroup has evolved more recently than D. virlis, prompting scientists to investigate the reason for a change from a euchromatic to heterochromatic dot chromosome. Figure 8. Blastn results indicating a section of a suspected gene sequence within fosmid13 after being run through a D. melanogaster gene database. The query sequence represents the D. melanogaster gene location within the chromosome, and the subject sequence is the suspected D. erecta gene. The sequences are 96% matches to one another, with an example of a mismatched base pair circled in red. Figure 2. Genome shotgun sequencing pathway indicating that gaps in the sequence of the assembly can form where no fosmids are recorded across a given area. Figure 7a. Visual representation of the Genscan program predicted genes from the annotation project containing fosmid13. Genscan has predicted 5 total genes within the fosmid, three in the plus reading frame and two in the minus reading frame. Figure 7b. A more specific read-out of predicted genes provided by the Genscan program is described. Contained within are predicted exon/intron boundaries are shown as well as relevant sequences such as the promoter region and poly-A tails. FINISHING A multi-step process used to piece together a complete and flawless DNA sequence GOALS: To eliminate any gaps, correct and improve low-quality regions, high-quality discrepancies between bases, single subclones, or single chemistry for sequencing reads PROCEDURE: * Select DNA fosmids of approximately 40 kb in length from an online database * Analyze these sequences by using the programs Consed and Phred/Phrap * Look at the gaps present in the fosmid and the overall quality of the sequence (Fig. 3) * Correct high-quality discrepancies between base pairs when enough evidence from other reads is present (Fig. 4) * Call for reads to solve more complex problems such as gaps or low-quality areas that have no relevant data present (Fig. 5) * Check for bacterial contaminants to be present with “BLAST” * Continue to annotation with the final finishing product is complete and has a high-quality DNA in comparison to the consensus (Fig. 6) Figure 9. A summary of the actual genes located within fosmid13 is shown. Notice that five genes were originally predicted (Fig. 7), however, only two genes were actually found to be present within fosmid13. Figure 3. A screenshot from the program Consed of the original assembly view of finishing project 120-D14 is shown. The DNA contig labeled 3 is a sequence approximately 29 kb in length, while contig 2 is roughly 12 kb in length. One of the primary duties of a finisher is to bridge the gap between these two contigs in order to create a single continuous contig roughly 41 kb long. The green lines spanning the assembly represent the amount of coverage over the area as well as relative quality of the data present. The triangle lines represent forward and reverse pairs, and the orange and black boxes show tandem and inverse repeats. Additional reads may be required to increase the quality of certain areas to an acceptable level. CONCLUSION The implementation of the Genomics Education Partnership at Longwood University was successful. At Longwood University a total of five annotation and four finishing projects were completed and submitted during my two-semester involvement with this project. The finishing of D. mojavensis and annotation of D. virilis has since been completed. Although, we did start with the annotation of D. erecta, this current year, the GEP had added to its research new drosophila species. Thus, the target organism for finishing is D. grimshawi while for annotationis D. mojavensis as well as to complete the remaining fosmids of D. erecta. The data obtained through the completion of these projects will go a long way in assisting upper-level researchers in determining the evolutionary transition from a euchromatic to heterochromatic dot chromosome. The GEP provides a unique educational experience, allowing students to be involved in a project that requires collaboration with other students and faculty spread across the country. Figure 5. Two separate sets of reads called for project 120-D14 are described. The column entitled Oligo Sequence shows the selected DNA primer sequence from which the read will originate from. Primers are selected 70-100 bases from the problem area and are oriented to span the region in question. Through use of these reads the two contigs were joined and all low-quality, single subclone/single chemistry areas were remedied. Figure 4. An example of a high quality discrepancy between base pairs is given in this figure. Each row represents a separate read over the target area. While all reads are of equal quality at that particular base (13,533 of the consensus sequence), some call it as a cytosine and others as a thymine. This discrepancy is an example of a polymorphism and can not be manually corrected by the finisher. Instead the finisher tags the area so that more detailed research can be conducted later on. REFERENCES GEP Homepage: http://gep.wustl.edu/ NCBI BLAST Search Engine: http://blast.ncbi.nlm.nih.gov/Blast.cgi FlyBase: http://flybase.org/ UCSC Genome Browser: http://genome.ucsc.edu/ RepeatMasker: http://www.repeatmasker.org/ Genscan: http://www.genscan.com/ ACKNOWLEDGMENTS GEP Program Director: Sarah C.R. Elgin Technical Director: Chris Shaffer Chief Technical/Teaching Assistant: Wilson Leung Sponsored by Washington University at St. Louis and HHMI GEP members and partner 06-07-08 and their students and institutions Biology 425 students, spring class 2008 at Longwood University Figure 6. Assembly view of project 120-D14, obtained upon project completion, showing that the initial gap has been bridged, and all other errors corrected. The fosmid will continue on to annotation from this point.






