Download

1 / 62
630 likes | 928 Views
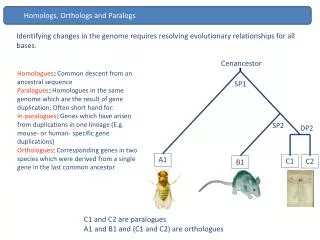
Paralogs. Inbal Yanover Reading Group in Computational Molecular Biology. Homologs. Orthologs : Homologous sequences are orthologous if they were separated by a speciation event Paralogs : paralogous if they were separated by a gene duplication event. Genomic duplication. Can involve :

E N D
Paralogs Inbal Yanover Reading Group in Computational Molecular Biology
Homologs • Orthologs: Homologous sequences are orthologous if they were separated by a speciation event • Paralogs: paralogous if they were separated by a gene duplication event
Genomic duplication Can involve: Individual genes • Genomic segments • Whole genome duplication (WGD) Gene duplication has a major role in evolution.
Whole genome duplication • Large scale adaptation • Polyploidy instability • Back to stability: • gene loss • mutation • genomic rearrangements
Fate of duplicated genes Find specialized ‘niche’: • Localization • Temporal expression • Expression level Another classification: • Sub – functionalization • Neo – functionalization (lowest probability) • Non – functionalization (70%)
First article Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae Kellis M, Birren BW, Lander ES. Nature. Apr 2004.
Main ideas • S. Cerevisiae genome arose from ancient whole-genome duplication of K. waltii • Analyzing post duplication divergence of paralogs
Expected signature for genome duplication: • After duplication, usually, one paralog would be lost (random local deletions) • Both copies will be retained only if they acquire distinct functions • Eventually: a few paralog genes in the same order and same orientation • Those regions should be short since chromosomal rearrangements will disrupt gene order over time
Model for WGD followed by massive gene loss Common ancestor
Proving existence of an ancient WGD • Look for a species (Y) in the lineage of S.cerevisiae (S). • Y and S should have 1:2 mapping and: • Nearly every region in Y would correspond to 2 regions in S (‘sister region’). • Each sister region in S would contain an ordered subsequence of the genes in Y. • Each sister region in S would contain ~half of Y genes. • Together, the two sister region account for nearly all Y’s genes. • Every region of S would correspond to one region in Y.
Y = K. Waltii • Sequencing and assembling into 8 complete chromosomes (16 in S. cerevisiae). • 5,230 likely protein-coding genes (5,714 genes in S. cerevisiae). • 7% of it’s genes shows no protein similarity to S. Cerevisiae • Identifying orthologs regions: • Matching genes (based on protein similarity) • Regions with numerous matching genes in the same order. • Most local regions in K. waltii mapped to two regions in S. cerevisiae. • Each of those regions matched subset of K. waltii genes.
Quantify observations DCS – Doubly Conserved Synteny: maximal regions in K. Waltii that map across their entire length to two distinct regions in S. cerevisiae.
Results • 253 DCS blocks containing most of both genomes. (75% of K. waltii genes and 81% of S. cerevisiae genes) • DCS blocks tile 85% of each K. waltii chromosome -> as expected in WGD • Typical DCS block: • 27 genes. • Separated by small segments (~3 genes), that match one conserved region in S. cerevisiae.
Duplicate mapping of centromers Note: no paralogs here !
Duplicated blocks in S. cerevisiae • Using the DCS blocks: define 253 sister regions in S. cerevisiae. • Many of those could not be recognized without K. waltii mediation.
Conclusion WGD event occurred in the Saccharomyces lineage after the divergence from K. waltii.
Pattern of gene loss • Number of chromosomes was doubled. • Despite WGD, current S. cerevisiae genome: • 13% larger than K. waltii genome. • 10% more genes. • Gene loss: • large segmental deletions <-> individual gene deletions. • Balanced between two paralogs <-> act primarily on one of them. • Analysis of DCS blocks show: • average size of lost segment: 2 genes. • average balance: 43%-57%.
Two models – what happens after duplication event • One copy preserves original function while the other one is free to diverge (Ohno) • Both copies would diverge more rapidly and acquire new functions
Evolutionary analysis Study the evolution of the 457 gene pairs that arose by WGD: • Use synteny to distinguish them from pairs which arose by local duplication events. • Compute divergence rates for them, using sequences of K. waltii, S. cerevisiae and S. bayanus. (both amino acid and nucleotides).
Results • 17% of gene pairs (76 of 457) showed acceleratedprotein evolution relative to K. waltii. • In 95% of them, accelerated evolution was confined to only one paralog • Supports Ohno’s model: one paralog retains ancestral function, the other one gains a derived function
Ancestral <-> derived paralogs • 115 gene pairs consisting of one paralog which has evolved >50% faster than the other. • Often, derived paralogs are specialized in: • Cellular localization (Acc1 - Hfa1) • Temporal expression (Skt5 – Shc1)
Ancestral <-> derived paralogs, cont. • Functional distinction confirmed with knockout experiments (in rich medium) of all 115 genes: • Deletion of ancestral paralog was lethal in 18%. • Deletion of derived paralog was never lethal. • Explanation: • Derived paralog is not essential under this conditions. • Ancestral paralogue compensate. (but not vice versa)
more results • 60 of the 457 pairs (13%) showed decelerated proteinevolution. • Including highly constrained proteins: • ribosomal proteins (25) • Histone proteins (2) • Translation factors (4) • In 90% of them both paralogs were very similar (98% amino acid identity versus 55% for all pairs)
However… • ~70% of the gene pairs had neither accelerated protein evolution nor decelerated evolution (321/457) • Possible explanations: • Too strict criteria • Divergence in regulatory regions will not be seen here. • Sometimes it’s nice to have two copies.
summary • S. cerevisiae arose from an ancient WGD. • Massive loss of ~90% of duplicated genes in small deletions. • Preserving at least one copy of each ancestral gene. • divergence of paralogs: • Accelerated evolution (17%) • Derived genes tend to be specialized in function, expression level and localization. • Derived genes tend to lose essential aspects of their ancestral function.
Second article Transcription control reprogramming in genetic backup circuits. Kafri R, Bar-Even A, Pilpel Y. Nat Genet. Mar 2005.
Introduction • Severe mutationsoften don’t result in abnormal phenotype • Partially ascribed to redundant paralogs, that provide backup to each other in case of mutation • Suggested mechanism: transcriptional reprogramming
Definitions • Working on S. cerevisiae. • Paralog pairs defined by BLASTing their DNA sequences. • Dispensable genes = non essential.
Expression parameters • For each pair of paralog: • Calculate 40 correlation coefficients of mRNA expression. • Define: mean expression similarity <= mean. • Define: partial co regulation (PCoR) <= standard deviation.
Summary of observations + : backup enabled
In close paralogs: • Backup increases with co-expression. Close paralogs • Backup increases with co-expression. • Similar sequences: • Similar expression • Enable backup
Remote paralogs • Backup is optimal in non-co expressed pairs. • co-expression (little backup): • interaction • sub-functionalization
Suggestion for backup mechanism • A, B - genes which are expressed differently. • Upon mutation in A: expression of gene B is reprogrammed. • Result: wild type expression profile of A.
Experimental verifier: reprogramming in Acs1/Acs2 Wild-type D Acs2 Glucose Glucose Acs2 Acs1 Acs1 Acs1 Acs2
What is the mechanism enabling this change? • Suggestion: backup occurs among paralogs with partially co regulation. • Enable switching from different expression profile to similar one. • Observation: PCoR predicts backup.
Partial motif content overlap is optimal for backup 1 |m1 ∩ m2| Backup measure O= 0.8 Proportion of dispensable genes |m1 U m2| 0.6 0 0.2 0.4 0.6 Motif content overlap (O)
suggestion • Unique motifs -> different expression level. • Shared motifs -> enable responding to the same conditions. Hypothesis: PCoR underlies reprogramming and backup.
In high PCoR paralogs one gene is upregulated when other is deleted 10 9 8 7 6 Fold change (Hughes et al. Cell 2000) 5 4 3 2 1 0 <0.35 0.35 – 0.45 >0.45 Partial co-regulation (predicted backup capacity)
M1 E1 G1 T E2 G2 M2 What controls reprogramming? • Kinetic model: G1, G2 – paralog genes. E1, E2 – their products. T – TF which is generated by M1 and has binding site in both genes.
Conclusions • In remote paralogs: Genes which express differently but has partial common regulation tends to backup each other. • In close paralogs: Backup increases with co-expression.
Third article Gene regulatory network growth by duplication Teichmann SA, Babu MM. Nat Genet. May, 2004
Main questions • What is the role of gene duplication in regulatory network evolution? • Determine the extent to which duplicated genes inherit interactions from their ancestors. • Describe possible mechanisms which leads to the formation of a new interaction.
Basic unit of gene regulation • Transcription factor • DNA binding site • Target gene (or transcription unit) Complex network: • 1 gene is regulated by few transcription factors. • 1 transcription factor controls more than one gene. Transcription factor Target gene
Gene regulatory network in E. coli Gene regulatory network in Yeast 795 proteins (121 TFs + 674 TGs) 1423 interactions 477 proteins (109 TFs + 368 TGs) 901 interactions Guelzim et.al. Nat. Gen. (2002) Shen-orr et.al. Nat. Gen. (2002) and RegulonDB Research subjects E. Coli and yeast known regulatory networks: > 100 transcription factors regulate several hundreds genes.
Duplication (reminder) • duplication event: • Inherit regulatory interaction • Lose regulatory interaction • Also, a new interaction may arise.
Homology detecting • structural protein homology • Detects more distant relationships than sequence • > 65% of the genes are the result of gene duplication • Same domain architecture -> common ancestor.
Duplication of transcription factor Loss and gain Inheritance Transcription factor Duplication of TF Target gene