Download

1 / 34
370 likes | 708 Views
Genotype Imputation. Dan Evans devans@psg.ucsf.edu California Pacific Medical Center Research Institute. Outline. Overview Elements of a Hidden Markov Model (HMM) Methods used by MACH Method comparison with IMPUTEv2 Implementation with MACH Implementation with IMPUTEv2

E N D
Genotype Imputation Dan Evans devans@psg.ucsf.edu California Pacific Medical Center Research Institute
Outline • Overview • Elements of a Hidden Markov Model (HMM) • Methods used by MACH • Method comparison with IMPUTEv2 • Implementation with MACH • Implementation with IMPUTEv2 • Software evaluation
Impute missing genotypes Li et al., Annu Rev Genomics Hum Genet 2009
Benefits of imputation • Expanded set of SNPs tested for association • Facilitate meta-analysis among studies using different genotyping arrays Marchini et al., Nature Genetics 2007
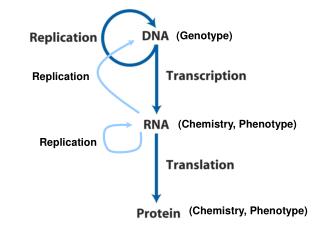
Imputation steps • Phase study genotypes • Impute missing genotypes from phased haplotypes Phase 1 M1 M2 M3 M4 ID1 G - - - G - - - T - - - A ID1 G - - - A - - - C - - - A Phase 2 M1 M2 M3 M4 ID1 G - - - A- - - T - - - A ID1 G - - - G- - - C - - - A
Phasing genome-wide • EM algorithm – treats all possible haplotype configurations as equally likely a priori • Computational constraints when markers > 10 • Hidden Markov Models – new haplotypes derived from older haplotypes by mutation and recombination • Limits the possible haplotype configurations
Outline • Overview • Elements of a Hidden Markov Model (HMM) • Methods used by MACH • Method comparison with IMPUTEv2 • Implementation with MACH • Implementation with IMPUTEv2 • Software evaluation
Elements of a Hidden Markov Model (HMM) • Probabilistic model for sequence annotation – identify the 5’ splice site • Exons, splice sites, and introns have different base composition • 3 states • Each state has emission probabilities • Each state has transition probabilities path = π Path is a Markov chain Eddy, Nature Biotechnology 2004
Probability model of sequence • Sequence x1 … xL, ith symbol xi • transition between different states in the path • emission – probability that symbol b is seen at position i when the ith state in the path is k • Joint probability of sequence and path Durbin et al., Biological sequence analysis, 1998 start transition emission transition
What if state path, emission probabilities and transition probabilities are unknown? • Dynamic programming algorithms to determine path • Viterbi algorithm • Forward – backward algorithm • Baum-Welch algorithm to estimate transition and emission probabilities
Forward algorithm • Probability of observed sequence up to and including xi, given statei = k Sum over all states At each position transition emission
Backward algorithm • Probability of observed sequence starting from the end and working backwards: Start at end Sum over all states at each position
Posterior state probabilities • Want to know probability of state k at position i when the emitted sequence is known • Posterior probability General multiplication rule Divide both sides by P(x) From posterior probability, can take most probable state, or apply function on states multiplied by posterior prob
Baum-Welch algorithm • Initial guess at transition () and emission probabilities () • Forward-backward to find posterior probabilities of states in path • Use posterior probabilities at each state to estimate new and • Iterate steps 2 and 3 until stopping criteria (small difference in log likelihood) Version of EM algorithm
Outline • Overview • Elements of a Hidden Markov Model (HMM) • Methods used by MACH • Method comparison with IMPUTEv2 • Implementation with MACH • Implementation with IMPUTEv2 • Software evaluation
MACH Haplotyping with HMM • Hidden – sequence of mosaic states S that emit the observed genotypes • Transition probabilities – recombination events • Emission probabilities – mutation, error Li et al., Genet Epidemiol 2010 start transition emission transition
MACH Path estimation • Forward – backward algorithm to estimate path • Update transition and emission probabilities with each estimated path, Baum algorithm • Rounds is the number of updates, 20 is suggested to estimate path and parameters
MACH genotype imputation • HMM again, but this time include reference haplotypes • count frequency that genotype was sampled at each position across iterations • Most probable genotype sampled most often • Expected number of allele counts (dosage) = 2*hom counts + het counts/# samples
MACH imputation quality measures • Quality of genotype = proportion of iterations where the final imputed genotype was selected • Quality of marker = genotype quality score averaged across all individuals • r2 = observed/expected variance of genotype scores • p=mean(g)/2 • Var(g)/[2*p*(1-p)]
Outline • Overview • Elements of a Hidden Markov Model (HMM) • Methods used by MACH • Method comparison with IMPUTEv2 • Implementation with MACH • Implementation with IMPUTEv2 • Software evaluation
IMPUTEv2 vs MACH • Transmission and emission probabilities • IMPUTEv2 uses fixed values for these parameters. Emission probability is constant assuming a uniform mutation rate. Transmission probability from the fine-scaled recombination map of human genome. • MACH estimates these parameters using Baum-Welch algorithm
IMPUTEv2 vs MACH • Potential states • IMPUTEv2 considers study and reference haplotypes • Reduces complexity using Hamming distance to select genetically more similar haplotypes • Can accommodate large reference panels • MACH randomly selects 200 haplotypes, doesn’t leverage all haplotypes
Outline • Overview • Elements of a Hidden Markov Model (HMM) • Methods used by MACH • Method comparison with IMPUTEv2 • Implementation with MACH • Implementation with IMPUTEv2 • Software evaluation
MACH ./mach1 \ -d ../examples/sample.dat \ -p ../examples/sample.ped\ -h ../examples/hapmap.haplos\ -s ../examples/hapmap.snps\ --rounds 50 \ #number of iterations --states 200 \ #number of haplotypes to sample --dosage \ #output dosage, not best genotypes --prefix ../output/test \ > ../output/dosage.log
Outline • Overview • Elements of a Hidden Markov Model (HMM) • Methods used by MACH • Method comparison with IMPUTEv2 • Implementation with MACH • Implementation with IMPUTEv2 • Software evaluation
IMPUTEv2 ./impute2 \ -m ./Example/example.chr22.map \ ##recombination map -h ./Example/example.chr22.1kG.haps \ ##reference haplotypes -l ./Example/example.chr22.1kG.legend \ ##SNP annotation for ref haplo -g ./Example/example.chr22.study.gens \ ##study genotypes -strand_g ./Example/example.chr22.study.strand \ ##study SNP strand -int 20.4e6 20.5e6 \ ##genomic interval -Ne 20000 \ ##effective population size, ##scales recombination rates -o ./Example/example.chr22.one.phased.impute2
---------------- Run parameters ---------------- reference haplotypes : 112 [Panel 0] study individuals : 250 [Panel 2] sequence interval : [20400000,20500000] buffer : 250 kb Ne : 20000 input call thresh : 0.900 #genotypes with P<0.9 are missing burn-in MCMC iterations : 10 #forward-backward that don’t contribute to imputation probabilities total MCMC iterations : 30 (20 used for inference) HMM states for phasing : 80 [Panel 2] HMM states for imputation : 112 [Panel 0->2] #make this large
Outline • Overview • Elements of a Hidden Markov Model (HMM) • Methods used by MACH • Method comparison with IMPUTEv2 • Implementation with MACH • Implementation with IMPUTEv2 • Software evaluation
Pre-phasing • Reference panels updated frequently • Phase study haplotypes with SHAPEIT2 • Impute ungenotyped SNPs with IMPUTEv2