Download

1 / 33
330 likes | 638 Views
RNA functions, structure and Phylogenetics. RNA functions. Storage/transfer of genetic information Genomes many viruses have RNA genomes single-stranded (ssRNA) e.g., retroviruses (HIV) double-stranded (dsRNA) Transfer of genetic information mRNA = "coding RNA" - encodes proteins.

E N D
RNA functions • Storage/transfer of genetic information • Genomes • many viruses have RNA genomes • single-stranded (ssRNA) • e.g., retroviruses (HIV) • double-stranded (dsRNA) • Transfer of genetic information • mRNA = "coding RNA" - encodes proteins
RNA functions • Structural • e.g., rRNA, which is a major structural component of ribosomes • BUT - its role is not just structural, also: • Catalytic • RNA in the ribosome has peptidyltransferase activity • Enzymatic activity responsible for peptide bond formation between amino acids in growing peptide chain • Also, many small RNAs are enzymes "ribozymes“ • Regulatory • Recently discovered important new roles for RNAs • In normal cells: • in "defense" - esp. in plants • in normal development • e.g., siRNAs, miRNA
RNA types & functions L Samaraweera 2005
Outline RNA Structure • RNA primary structure • RNA secondary structure & prediction • RNA tertiary structure & prediction
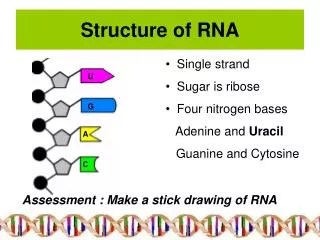
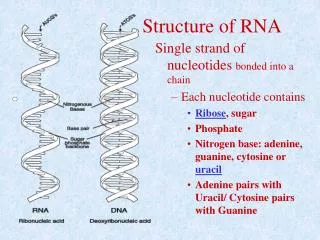
Primary structure • 5’ to 3’ list of covalently linked nucleotides, named by the attached base • Commonly represented by a string S over the alphabet Σ={A,C,G,U}
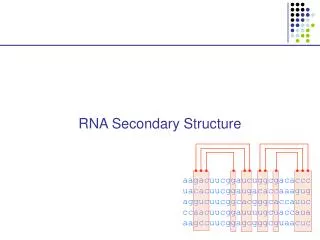
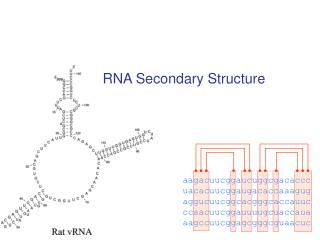
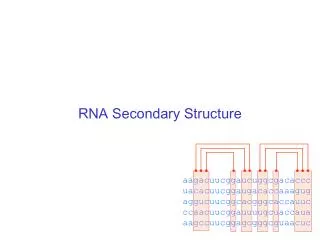
Secondary Structure Listof base pairs, denoted by i•j for a pairing between the i-th and j-th Nucleotides, ri and rj, where i<j by convention. Helices are inferred when two or more base pairs occur adjacent to one another Single stranded bases within a stem are called a bulge of bulge loop if the single stranded bases are on only one side of the stem. If single stranded bases interrupt both sides of a stem, they are called an internal (interior) loop.
RNA secondary structure representation ..(((.(((......))).((((((....)))).))....))) AGCUACGGAGCGAUCUCCGAGCUUUCGAGAAAGCCUCUAUUAGC
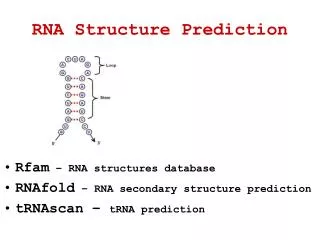
RNA structure prediction • Twoprimary methods for ab initio RNA secondary • structure prediction: • Co-variation analysis (comparative sequence analysis) • . Takes into account conserved patterns of base pairs during • evolution (more than 2 sequences) • Minimum free-energy method • . Determine structure of complementary regions that are • energetically stable
RNA folding: Dynamic Programming There are only four possible ways that a secondary structure of nested base pair can be constructed on a RNA strand from position i to j: • i is unpaired, added on to • a structure for i+1…j • S(i,j) = S(i+1,j) • j is unpaired, added on to • a structure for i…j-1 • S(i,j) = S(i,j-1)
RNA folding: Dynamic Programming • i j paired, but not to each other; • the structure for i…j adds together • structures for 2 sub regions, • i…k and k+1…j • S(i,j) = max {S(i,k)+S(k+1,j)} • i j paired, added on to • a structure for i+1…j-1 • S(i,j) = S(i+1,j-1)+e(ri,rj) i<k<j
RNA folding: Dynamic Programming Since there are only four cases, the optimal score S(i,j) is just the maximum of the four possibilities: To compute this efficiently, we need to make sure that the scores for the smaller sub-regions have already been calculated
Other methods • Base pair partition functions • Calculate energy of all configurations • Lowest energy is the prediction • Statistical sampling • Randomly generating structure with probability distribution = energy function distribution • This makes it more likely that lowest energy structure is found • Sub-optimal sampling
RNA tertiary structure (interactions) In addition to secondary structural interactions in RNA, there are also tertiary interactions, including: (A) pseudoknots, (B) kissing hairpins and (C) hairpin-bulge contact. Pseudoknot Kissing hairpins Hairpin-bulge Do not obey “parentheses rule”
Useful web sites on RNA • Comparative RNA web site http://www.rna.icmb.utexas.edu/ • RNA world http://www.imb-jena.de/RNA.html • RNA page by Michael Suker http://www.bioinfo.rpi.edu/~zukerm/rna/ • RNA structure database http://www.rnabase.org/ http://ndbserver.rutgers.edu/ (nucleic acid database) http://prion.bchs.uh.edu/bp_type/ (non canonical bases) • RNA structure classification http://scor.berkeley.edu/ • RNA visualisation http://ndbserver.rutgers.edu/services/download/index.html#rnaview http://rutchem.rutgers.edu/~xiangjun/3DNA/
Phylogenetics • Phylogenetics is the branch of biology that deals with evolutionary relatedness • Phylogenetics = studying or estimating the evolutionary relationships among organisms • Phylogenetics on sequence data is an attempt to reconstruct the evolutionary history of those sequences • Relationships between individual sequences are not necessarily the same as those between the organisms they are found in • The ultimate goal is to be able to use sequence data from many sequences to give information about phylogenetic history of organisms
History • Darwin (1872) Included a tree diagram in On the Origin of Species • Haeckel (1874) “Ontogeny recapitulates phylogeny” • Phenetics (Sneath, Sokal, Rohlf) Common ancestry cannot be inferred so organisms should be grouped by overall similarity Distance-based methods
Phylogenetic tree • Node = ancestral taxa • Root = common ancestor of all taxa on the tree • Clade = group of taxa and their common ancestor • Branch length may be scaled to represent time, substitutions • Nodes may be rotated without a change in meaning • May include extant and extinct taxa
Phylogenetic tree Phylogenetic relationships usually depicted as trees, with branches representing ancestors of “children”; the bottom of the tree (individual organisms) are leaves. Individual branch points are nodes. C A D time B A B C D A rooted tree An unrooted tree time?
Characteristics of the tree • We will only consider binary trees: edges split only into two branches (daughter edges) • rooted trees have an explicit ancestor; the direction of time is explicit in these trees • unrooted trees do not have an explicit ancestor; the direction of time is undetermined in such trees
Tree Construction Several methods: • Distance-based or Clustering methods • Parsimony • Likelihood • Bayesian
Types of phylogenetic analysis methods • Phenetic: trees are constructed based on observed characteristics, not on evolutionary history • Cladistic: trees are constructed based on fitting observed characteristics to some model of evolutionary history Distance methods Parsimony and Maximum Likelihood methods
Distance matrix methods • Create a matrix of the distance between each pair of organisms and create a tree that matches the distances as closely as possible • Pairwise distance, Least squares, minimum evolution, UPGMA, neighbor-joining methods • Distance scoring matrices for amino acid sequences
Parsimony • Parsimony methods are based on the idea that the most probable evolutionary pathway is the one that requires the smallest number of changes from some ancestral state • For sequences, this implies treating each position separately and finding the minimal number of substitutions at each position • Convergent evolution, parallel evolution, & reversals ==> homoplasy • Susceptible to long-branch attraction (due to high probability of convergent evolution)
Maximum Likelihood • Search among all possible trees for the tree with the highest probability or likelihood of producing our data given a particular model of evolution • Maximum likelihood reconstructs a tree according to an explicit model of evolution. • But, such models must be simple, because the method is computationally intensive
Bayesian Analysis • Similar to Likelihood, but it searches among all possible trees to find the tree with the highest likelihood or probability of occurring given our data
Models of evolution Vary in the number and type of parameters to be optimized: • base frequencies • substitution rates • transition/transversion ratios • Separate models of evolution in individual nucleotides, codons, or amino acids
How many possible trees?!? OrganismsTrees 1 1 2 1 3 3 4 15 5 105 6 945 7 10,395 8 135,135 9 2,027,025 10 34,459,425 15 213,458,046,676,875 30 4.9518E38 50 2.75292E76 Searching for the optimal tree…
Support for phylogenetic methods • Bacteriophage T7 (Hillis et al. 1992): Picked correct tree topology out of 135,135 possibilities using 5 different methods. Branch lengths varied. • Lab mice (Atchely & Fitch 1991): “Almost perfectly” identified the known genealogical relationships among 24 strains of mice.
Assessing trees • The bootstrap: randomly sample all positions (columns in an alignment) with replacement -- meaning some columns can be repeated -- but conserving the number of positions; build a large dataset of these randomized samples
The bootstrap sampling • Then use your method (distance, parsimony, likelihood) to generate another tree • Do this a thousand or so times • Note that if the assumptions the method is based on hold, you should always get the same tree from the bootstrapped alignments as you did originally • The frequency of some feature of your phylogeny in the bootstrapped set gives some measure of the confidence you can have for this feature
Phylogeny programs • PHYLIP- one of the earliest (1980), freely distributed, parsimony, maximum likelihood, and distance matrix methods • PAUP*- probably most widely used, parsimony, likelihood, and distance matrix methods, more features than PHYLIP • MacClade, MEGA, PAML, TREE-PUZZLE, DAMBE, NONA, TNT, many others
Orthologs vs. Paralogs • When comparing gene sequences, it is important to distinguish between identical vs. merely similar genes in different organisms. • Orthologs are homologous genes in different species with analogous functions. • Paralogs are similar genes that are the result of a gene duplication. • A phylogeny that includes both orthologs and paralogs is likely to be incorrect. • Sometimes phylogenetic analysis is the best way to determine if a new gene is an ortholog or paralog to other known genes.