Download
1 / 17
• 170 likes • 259 Views
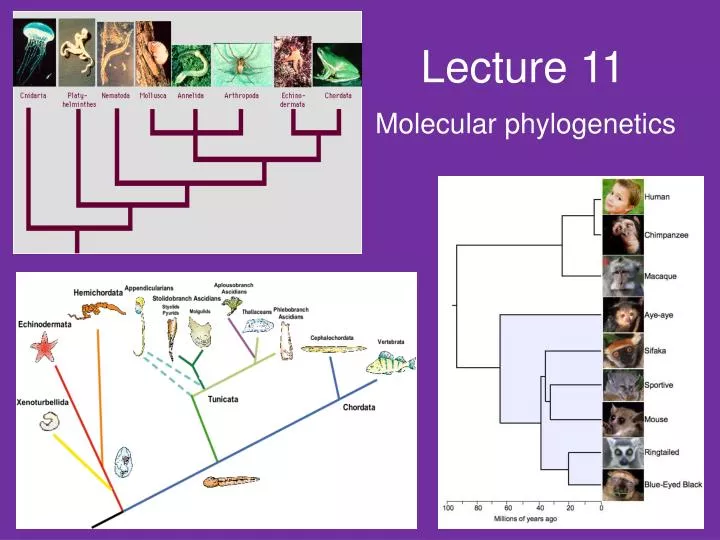
Lecture 11. Molecular phylogenetics. The goal phylogeny is simply to reconstruct the historical relationships between a group of taxa NB Phylogenetic trees are hypotheses

E N D
Lecture 11 Molecular phylogenetics
The goal phylogeny is simply to reconstruct the historical relationships between a group of taxa • NBPhylogenetic trees are hypotheses • the tree may have very strong support, or it may have very little support - a large number of characters supporting a specific topology • Weak support for a topology may arise due to – many possible alternatives which cannot be excluded
NB Gene trees are not the same as species trees • species trees illustrate the evolutionary histories of a group of related species • i.e., species trees record the details of speciation for the group • gene trees show the evolutionary relationships among DNA sequences for a locus • gene trees may not be the same as species trees for one main reason – the existence of ancestral polymorphism. • if this ancestral polymorphism is lost in some taxa but not in others, then one sequence isolated in species A may be more closely related to one in species B than to any other conspecific sequence. • the gene tree will thus be different from the true species tree. • the best way to guarantee that this will no occur is to use information provided by multiple independent loci!
Terminology • Trees may be Rooted and Unrooted • By an Outgroup– Which is a taxon assumed to have diverged before the taxa studied • Topology – refers to the branching pattern • You can have internal and external branches • Any type of data can be used to reconstruct phylogenetic trees • Historically morphological data was used • Molecular data sources are: • Allozymes, immunological distance, DNA-DNA hybridization, Restriction site data, Amino acid sequences, DNA sequence data
Characters • Characters may be binary (i.e., presence or absence of an isozyme allele) or multistate (i.e., ACGT) • characters may also be ordered or unordered • when characters are ordered a certain directionality is implied among changes
The important stuff • Two important assumptions about the characters used to build trees: • 1. the characters are independent • 2. the characters are homologous • a homologous character is one shared by two species because it was inherited from a common ancestor • homoplasy
How do we construct trees? • There are four major types of phylogenetic methods: 1. Distance methods (e.g. UPGMA, NJ) 2. Maximum parsimony methods (MP) 3. Maximum likelihood methods (ML) 4. Bayesian tree-building methods (such as that used by the computer program MrBayes) • MP and ML are both called “cladistic” methods
DistanceMethods • The general strategy behind distance methods is to cluster taxa (or OTUs) so that the most similar ones are found close together in the tree • This strategy is called a pheneticapproach • The best tree, according to this approach, is to minimize the total distance among all taxa • The branch lengths in phenograms carry important information • The closer two taxa resemble one another the higher they are positioned in the phenogram
Maximum parsimony (MP) • according to the MP approach, the best tree minimizes the number of evolutionary steps (i.e., changes among characters) • this is the principle of parsimony - the least number of changes, required the better the tree • evolutionary change does not always obey laws of parsimony but it is a reasonable starting point • MP trees are based exclusively on synapomorphies • shared by two or more taxa that are derived from some ancestral state • essential to use one or more outgroups
Maximum Likelihood (ML) • given a certain model of base substitution and a specific tree, what is the probability of obtaining this set of DNA sequences? • this probability is estimated by a tree’s likelihood score • the best tree is that which has the highest likelihood, or probability of being produced • ML methods are computationally intensive and thus have not been easy to use until recently
How do you select the correct tree • Correlation between distance, MP, and ML topologies is found – Reconstruction is GOOD • Common approach is to use a statistical technique called bootstrapping • Bootstrapping procedure works by randomly re-sampling the nucleotide sequence data (with replacement), constructing a tree from this data and counting the number of times a particular branch is found out of say, 100, 500, or 1,000 replicate pseudosamples
So? • Trees based on genetic markers will accurately reflect evolutionary relationships only if the • rates of evolution are constant • markers are neutral • foundation population is monomorphic (or there are many independently inherited markers) • If starting population is polymorphic, fixation of different initial sequences in different lineages (termed lineage sorting) may lead to incorrect inferred phylogenies
Outbreeding depression • Outbreeding depression is the reduction in reproductive fitness resulting from crossing of populations • Occurs when: adaptation to a certain area occurred; crossing different sub-species
Diagnosing genetic problems 1. How large is the population (Ne)? 2. Has it experienced significant bottlenecks in the past? 3. Has it lost genetic diversity? 4. Is it suffering from inbreeding depression? 5. Is it genetically fragmented? • How do you recover a small population? • outbred (if available), or • inbred but genetically differentiated from the population to which they are being introduced • DL -Eldridge et al. 1999 – Black-footed Rock wallaby
Genetically viable populations • How large must populations be,to be genetically viable in the long term? • minimum viable population size (MVP) • the minimum size required to retain reproductive fitness and evolutionary potential over thousands of years
How large? • Three genetic components must be considered in answering this question: • Is the population size large enough to avoid inbreeding depression? • Is there sufficient genetic diversity for evolution to occur in response to environmental change? • Is the population large enough to avoid accumulating new deleterious mutations?