Download
1 / 47
480 likes | 640 Views
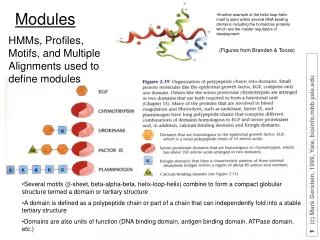
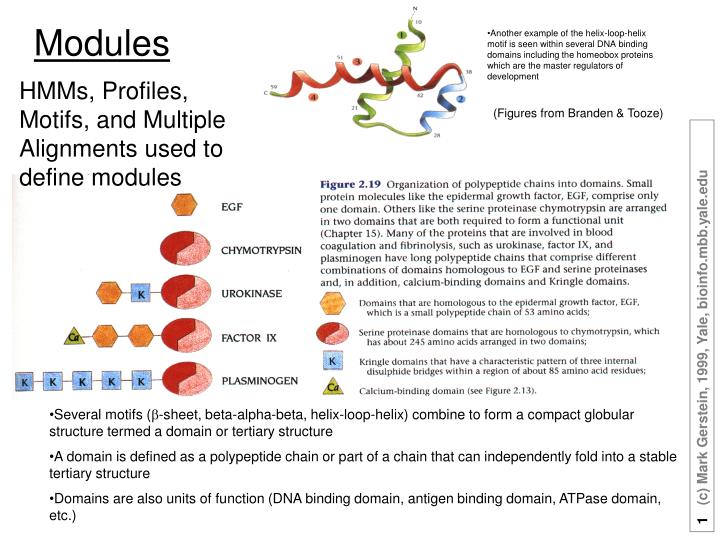
Another example of the helix-loop-helix motif is seen within several DNA binding domains including the homeobox proteins which are the master regulators of development. Modules. HMMs, Profiles, Motifs, and Multiple Alignments used to define modules. (Figures from Branden & Tooze).

E N D
Another example of the helix-loop-helix motif is seen within several DNA binding domains including the homeobox proteins which are the master regulators of development Modules HMMs, Profiles, Motifs, and Multiple Alignments used to define modules (Figures from Branden & Tooze) • Several motifs (b-sheet, beta-alpha-beta, helix-loop-helix) combine to form a compact globular structure termed a domain or tertiary structure • A domain is defined as a polypeptide chain or part of a chain that can independently fold into a stable tertiary structure • Domains are also units of function (DNA binding domain, antigen binding domain, ATPase domain, etc.)
COG 272, BRCT family P. Bork et al
Five Principal Fold Classes All a folds All b folds a + b folds a / b folds small irregular folds
SCOP - Protein Fold Hierarchy Class - 5 Fold - ~500 Superfamily - ~ 700 Family ~ 1000 Family - domains with common evolutionary origin
} “Twilight zone” Sequence Similarity May Miss Functional Homologies Which Can Be Detected by3D Structural Analysis Homologous 3D Structure Non-homologous 3D Structure % Sequence Identity Residues Aligned Adapted from Chris Sander
Structural Validation of Homology 19% Seq ID Z = 12.2 Adenylate Kinase Guanylate Kinase
Asp tRNA Synthetase Staphylococcal Nuclease CspA Gene 5 ssDNA Binding Protein Topoisomerase I CspB
What is Protein Geometry? • Coordinates (X, Y, Z’s) • Dihedral Angles • Assumes standard bond lengths and bond angles
Atom Position, XYZ triplets Other Aspects of Structure, Besides just Comparing Atom Positions Lines, Axes, Angles Surfaces, Volumes
Hb Automatic Alignment to Build Fold Library Alignment of Individual Structures Fusing into a Single Fold “Template” Mb Hb VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS-----HGSAQVKGHGKKVADALTNAV ||| .. | |.|| | . | . | | | | | | | .| .| || | || . Mb VLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRFKHLKTEAEMKASEDLKKHGVTVLTALGAIL Hb AHVD-DMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR------ | | . || | .. . .| .. | |..| . . | | . ||. Mb KK-KGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRHPGDFGADAQGAMNKALELFRKDIAAKYKELGYQG Elements: Domain definitions; Aligned structures, collecting together Non-homologous Sequences; Core annotation Previous work: Remington, Matthews ‘80; Taylor, Orengo ‘89, ‘94; Artymiuk, Rice, Willett ‘89; Sali, Blundell, ‘90; Vriend, Sander ‘91; Russell, Barton ‘92; Holm, Sander ‘93; Godzik, Skolnick ‘94; Gibrat, Madej, Bryant ‘96; Falicov, F Cohen, ‘96; Feng, Sippl ‘96; G Cohen ‘97; Singh & Brutlag, ‘98
Explain Concept of Distance Matrix on Blackboard N x N distance matrix N dimensional space Metric matrix Mij = Dij2 - Dio2 - Djo2 Eigenvectors of metric matrix Principal component analysis
Automatically Comparing Protein Structures Given 2 Structures (A & B), 2 Basic Comparison Operations 1 Given an alignment optimally SUPERIMPOSE A onto B Find Best R & T to move A onto B 2 Find an Alignment between A and B based on their 3D coordinates
RMS Superposition (2):Distance Betweenan Atom in 2 Structures
RMS Superposition (3):RMS Distance BetweenAligned Atoms in 2 Structures
RMS Superposition (4):Rigid-Body Rotation and Translationof One Structure (B)
RMS Superposition (5):Optimal Movement of One Structure to Minimize the RMS Methods of Solution: springs(F ~ kx) SVD Kabsch
Structural Alignment (1b) Make a Similarity Matrix(Generalized Similarity Matrix) • PAM(A,V) = 0.5 • Applies at every position • S(aa @ i, aa @ J) • Specific Matrix for each pair of residues i in protein 1 and J in protein 2 • Example is Y near N-term. matches any C-term. residue (Y at J=2) • S(i,J) • Doesn’t need to depend on a.a. identities at all! • Just need to make up a score for matching residue i in protein 1 with residue J in protein 2 i J
Structural Alignment (1c*)Similarity Matrixfor Structural Alignment • Structural Alignment • Similarity Matrix S(i,J) depends on the 3D coordinates of residues i and J • Distance between CA of i and J • M(i,j) = 100 / (5 + d2) • Threading • S(i,J) depends on the how well the amino acid at position i in protein 1 fits into the 3D structural environment at position J of protein 2
Alignment (2): Dynamic Programming,Start Computing the Sum Matrix new_value_cell(R,C) <= cell(R,C) { Old value, either 1 or 0 } + Max[ cell (R+1, C+1), { Diagonally Down, no gaps } cells(R+1, C+2 to C_max),{ Down a row, making col. gap } cells(R+2 to R_max, C+2) { Down a col., making row gap } ]
Alignment (5): Traceback Find Best Score (8) and Trace BackA B C N Y - R Q C L C R - P MA Y C - Y N R - C K C R B P
ACSQRP--LRV-SH -R SENCVA-SNKPQLVKLMTH VK DFCV- In Structural Alignment, Not Yet Done (Step 6*) • Use Alignment to LSQ Fit Structure B onto Structure A • However, movement of B will now change the Similarity Matrix • This Violates Fundamental Premise of Dynamic Programming • Way Residue at i is aligned can now affect previously optimal alignment of residues(from 1 to i-1)
How central idea of dynamic programming is violated in structural alignment
Structural Alignment (7*), Iterate Until Convergence 1 Compute Sim. Matrix 2 Align via Dyn. Prog. 3 RMS Fit Based on Alignment 4 Move Structure B 5 Re-compute Sim. Matrix 6 If changed from #1, GOTO #2
Some Similarities are Readily Apparent others are more Subtle Easy:Globins 125 res., ~1.5 Å Tricky:Ig C & V 85 res., ~3 Å Very Subtle: G3P-dehydro-genase, C-term. Domain >5 Å
Some Similarities are Readily Apparent others are more Subtle Easy:Globins 125 res., ~1.5 Å Tricky:Ig C & V 85 res., ~3 Å Very Subtle: G3P-dehydro-genase, C-term. Domain >5 Å
DALI: Protein Structure Comparison by Alignment of Distance MatricesL. Holm and C. Sander J. Mol. Biol. 233: 123 (1993) • Generate Ca-Ca distance matrix for each protein A and B • Decompose into elementary contact patterns; e.g. hexapeptide-hexapeptide submatrices • Systematic comparisons of all elementary contact patterns in the 2 distance matrices; similar contact patterns are stored in a “pair list” • Assemble pairs of contact patterns into larger consistent sets of pairs (alignments), maximizing the similarity score between these local structures • A Monte-Carlo algorithm is used to deal with the combinatorial complexity of building up alignments from contact patterns • Dali Z score - number of standard deviations away from mean pairwise similarity value
Structural Validation of Homology 19% Seq ID Z = 12.2 Adenylate Kinase Guanylate Kinase
Dali Domain DictionaryDeitman, Park, Notredame, Heger, Lappe, and Holm Nucleic Acids Res. 29: 5557 (2001) • Dali Domain Dictionary is a numerical taxonomy of all known domain structures in the PDB • Evolves from Dali / FSSP Database Holm & Sander, Nucl. Acid Res. 25: 231-234 (1997) • Dali Domain Dictionary Sept 2000 • 10,532 PDB enteries • 17,101 protein chains • 5 supersecondary structure motifs (attractors) • 1375 fold types • 2582 functional families • 3724 domain sequence families
Most proteins in biology have been produced by the duplication, divergence and recombination of the members of a small number of protein families. courtesy of C. Chothia
Cadherins courtesy of C. Chothia
A Global Representation of Protein Fold SpaceHou, Sims, Zhang, Kim, PNAS 100: 2386 - 2390 (2003) Database of 498 SCOP “Folds” or “Superfamilies” The overall pair-wise comparisons of 498 folds lead to a 498 x 498 matrix of similarity scores Sijs, where Sij is the alignment score between the ith and jth folds. An appropriate method for handling such data matrices as a whole is metric matrix distance geometry . We first convert the similarity score matrix [Sij] to a distance matrix [Dij] by using Dij = Smax - Sij, where Smax is the maximum similarity score among all pairs of folds. We then transform the distance matrix to a metric (or Gram) matrix [Mij] by using Mij = Dij2 - Dio2 - Djo2 where Di0, the distance between the ith fold and the geometric centroid of all N = 498 folds. The eigen values of the metric matrix define an orthogonal system of axes, called factors. These axes pass through the geometric centroid of the points representing all observed folds and correspond to a decreasing order of the amount of information each factor represents.
A Global Representation of Protein Fold SpaceHou, Sims, Zhang, Kim, PNAS 100: 2386 - 2390 (2003)