Download
1 / 1
10 likes | 91 Views
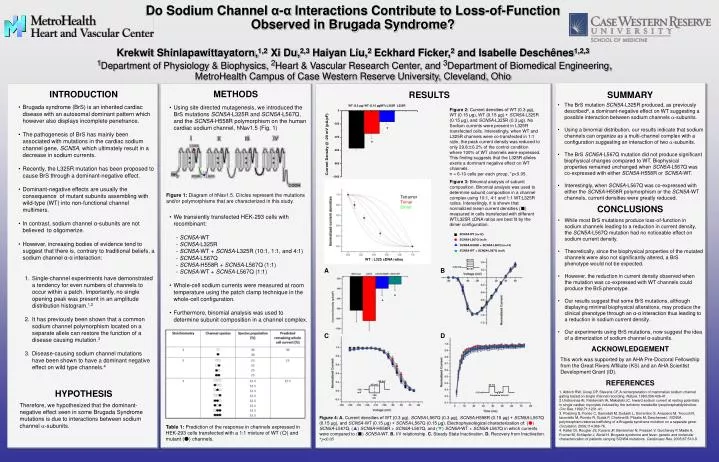
WT (0.3 µg). WT (0.15 µg). WT+L325R. L325R. *. *. SCN5A -WT (n=12). SCN5A -L567Q (n=9). -30 mV. SCN5A -H558R + SCN5A- L567Q (n=14). -65 mV. SCN5A -WT + SCN5A- L567Q (n=9). Tetramer Trimer Dimer. -120 mV. -140 mV. A. B. *. *. C. D. -30 mV. -120 mV. Interpulse Interval.

E N D
WT (0.3 µg) WT (0.15 µg) WT+L325R L325R * * SCN5A-WT (n=12) SCN5A-L567Q (n=9) -30 mV SCN5A-H558R + SCN5A-L567Q (n=14) -65 mV SCN5A-WT + SCN5A-L567Q (n=9) Tetramer Trimer Dimer -120 mV -140 mV A B * * C D -30 mV -120 mV Interpulse Interval Do Sodium Channel α-α Interactions Contribute to Loss-of-Function Observed in Brugada Syndrome?Krekwit Shinlapawittayatorn,1,2Xi Du,2,3 Haiyan Liu,2 Eckhard Ficker,2 and Isabelle Deschênes1,2,31Department of Physiology & Biophysics, 2Heart & Vascular Research Center, and 3Department of Biomedical Engineering, MetroHealth Campus of Case Western Reserve University, Cleveland, Ohio METHODS INTRODUCTION RESULTS SUMMARY • Brugada syndrome (BrS) is an inherited cardiac disease with an autosomal dominant pattern which however also displays incomplete penetrance. • The pathogenesis of BrS has mainly been associated with mutations in the cardiac sodium channel gene, SCN5A, which ultimately result in a decrease in sodium currents. • Recently, the L325R mutation has been proposed to cause BrS through a dominant-negative effect. • Dominant-negative effects are usually the consequence of mutant subunits assembling with wild-type (WT) into non-functional channel multimers. • In contrast, sodium channel α-subunits are not believed to oligomerize. • However, increasing bodies of evidence tend to suggest that there is, contrary to traditional beliefs, a sodium channel α-α interaction: • Single-channel experiments have demonstrated a tendency for even numbers of channels to occur within a patch. Importantly, no single opening peak was present in an amplitude distribution histogram.1,2 • It has previously been shown that a common sodium channel polymorphism located on a separate allele can restore the function of a disease causing mutation.3 • Disease-causing sodium channel mutations have been shown to have a dominant negative effect on wild type channels.4 • Using site directed mutagenesis, we introduced the BrS mutations SCN5A-L325R and SCN5A-L567Q,and the SCN5A-H558R polymorphism on the human cardiac sodium channel, hNav1.5 (Fig. 1) • We transiently transfected HEK-293 cells with recombinant: • - SCN5A-WT • - SCN5A-L325R • - SCN5A-WT + SCN5A-L325R (10:1, 1:1, and 4:1) • - SCN5A-L567Q • - SCN5A-H558R + SCN5A-L567Q (1:1) • - SCN5A-WT + SCN5A-L567Q (1:1) • Whole-cell sodium currents were measured at room temperature using the patch clamp technique in the whole-cell configuration. • Furthermore, binomial analysis was used to determine subunit composition in a channel complex. • The BrS mutation SCN5A-L325R produced, as previously described4, a dominant-negative effect on WT suggesting a possible interaction between sodium channels a-subunits. • Using a binomial distribution, our results indicate that sodium channels can organize as a multi-channel complex with a configuration suggesting an interaction of two a-subunits. • The BrS SCN5A-L567Q mutation did not produce significant biophysical changes compared to WT. Biophysical properties remained unchanged when SCN5A-L567Q was co-expressed with either SCN5A-H558R or SCN5A-WT. • Interestingly, when SCN5A-L567Q was co-expressed with either the SCN5A-H558R polymorphism or the SCN5A-WT channels, current densities were greatly reduced. Figure 2: Current densities of WT (0.3 μg), WT (0.15 μg), WT (0.15 μg) + SCN5A-L325R (0.15 μg), and SCN5A-L325R (0.3 μg). No Sodium currents were present in L325R transfected cells. Interestingly, when WT and L325R channels were co-transfected in 1:1 ratio, the peak current density was reduced to only 29.8±6.2% of the control condition where 100% of WT channels were expressed. This finding suggests that the L325R alleles exerts a dominant negative effect on WT channels. n = 6-10 cells per each group, *p<0.05. Figure 3: Binomial analysis of subunit composition. Binomial analysis was used to determine subunit composition in a channel complex using 10:1, 4:1 and 1:1 WT:L325R ratios. Interestingly, it is shown that normalized mean current densities (■) measured in cells transfected with different WT:L325R cDNA ratios are best fit by the dimer configuration. Figure 1: Diagram of hNav1.5. Circles represent the mutations and/or polymorphisms that are characterized in this study. CONCLUSIONS • While most BrS mutations produce loss-of-function in sodium channels leading to a reduction in current density, the SCN5A-L567Q mutation had no noticeable effect on sodium current density. • Theoretically, since the biophysical properties of the mutated channels were also not significantly altered, a BrS phenotype would not be expected. • However, the reduction in current density observed when the mutation was co-expressed with WT channels could produce the BrS phenotype. • Our results suggest that some BrS mutations, although displaying minimal biophysical alterations, may produce the clinical phenotype through an α-α interaction thus leading to a reduction in sodium current density. • Our experiments using BrS mutations, now suggest the idea of a dimerization of sodium channel α-subunits. +60 mV -120 mV -80 mV WT : L325 cDNA ratios ACKNOWLEDGEMENT This work was supported by an AHA Pre-Doctoral Fellowship from the Great Rivers Affiliate (KS) and an AHA Scientist Development Grant (ID). REFERENCES HYPOTHESIS 1. Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436-41. 2.Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231-41. 3. Poelzing S, Forleo C, Samodell M, Dudash L, Sorrentino S, Anaclerio M, Troccoli R, Iacoviello M, Romito R, Guida P, Chahine M, Pitzalis M, Deschenes I. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation. 2006;114:368-76. 4. Keller DI, Rougier JS, Kucera JP, Benammar N, Fressart V, Guicheney P, Madle A, Fromer M, Schlapfer J, Abriel H. Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res. 2005;67:510-9. Therefore, we hypothesized that the dominant-negative effect seen in some Brugada Syndrome mutations is due to interactions between sodium channel a-subunits. Figure 4: A. Current densities of WT (0.3 μg), SCN5A-L567Q (0.3 μg), SCN5A-H558R (0.15 μg) + SCN5A-L567Q (0.15 μg), and SCN5A-WT (0.15 μg) + SCN5A-L567Q (0.15 μg). Electrophysiological characterization of: (●) SCN5A-L567Q, (▲) SCN5A-H558R + SCN5A-L567Q, and (▼) SCN5A-WT + SCN5A-L567Q in which currents were compared to (■) SCN5A-WT. B. I/V relationship. C. Steady State Inactivation. D. Recovery from Inactivation. *p<0.05 Table 1: Prediction of the response in channels expressed in HEK-293 cells transfected with a 1:1 mixture of WT (○) and mutant (●) channels.
