Download
1 / 1
10 likes | 143 Views
Network and Pathway Based Analysis of Cancer Progression

E N D
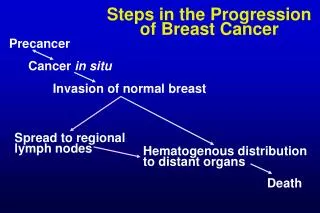
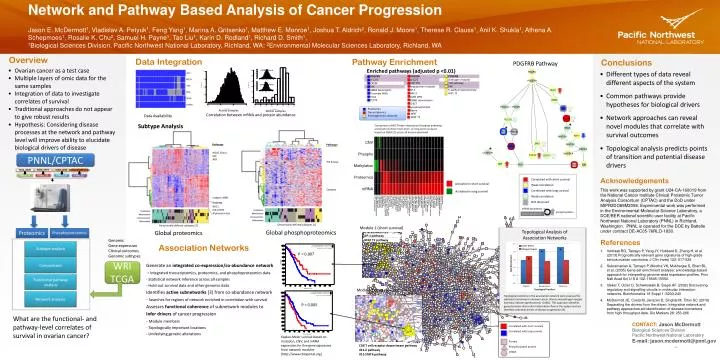
Network and Pathway Based Analysis of Cancer Progression Jason E. McDermott1,Vladislav A. Petyuk1, FengYang1, Marina A. Gritsenko1, Matthew E. Monroe1, Joshua T. Aldrich2, Ronald J. Moore1, Therese R. Clauss1, Anil K. Shukla1, Athena A. Schepmoes1, Rosalie K. Chu2, Samuel H. Payne1, Tao Liu1, Karin D. Rodland1, Richard D. Smith1, 1Biological Sciences Division, Pacific Northwest National Laboratory, Richland, WA; 2Environmental Molecular Sciences Laboratory, Richland, WA TCGA_XXXX TCGA_YYYY TCGA_ZZZZ Universal Reference iTRAQ 114 iTRAQ 115 iTRAQ 116 iTRAQ 117 PDGFRB IL-12/2 CXCR4 FAK AMB2 Neutrophils Thrombin PAR1 TXA2 TCPTP PDGFRB IL-12/2 CD8 TCR Angiopoietinreceptor AP-1 ARF-3 AVB3 OPN ERBB1 downstream IL-6/7 Lysophospholipid Netrin PDGFRB Androgen receptor TCR pathway FAK E-cadherin/keratinocyte HIF1 TF WNT NFAT TF Overview Data Integration Pathway Enrichment Conclusions PDGFRB Pathway Correlated with short survival • Ovarian cancer as a test case • Multiple layers of omic data for the same samples • Integration of data to investigate correlates of survival • Traditional approaches do not appear to give robust results • Hypothesis: Considering disease processes at the network and pathway level will improve ability to elucidate biological drivers of disease mRNA alone Weak correlation • Different types of data reveal different aspects of the system • Common pathways provide hypotheses for biological drivers • Network approaches can reveal novel modules that correlate with survival outcomes • Topological analysis predicts points of transition and potential disease drivers Correlated with long survival Proteomics Transcriptomics Proteogenomics aberrant Weak correlation protein alone Not observed mRNA abundance phosphorylation protein abundance Across samples Within samples Correlation between mRNA and protein abundance Data Availability * Subtype Analysis Comparison of NCI Protein Interaction Database pathways enriched in tumors from short- or long-term survivors based on GSEA [2] across all tumors examined. * Pathways Pathways * HDAC Class I E2F ATR PNNL/CPTAC PI3 Kinase Enriched pathways (adjusted p <0.01) Acknowledgements • This work was supported by grant U24-CA-160019 from the National Cancer Institute Clinical Proteomic Tumor Analysis Consortium (CPTAC) and the DoDunder MIPR2DO89M2058. Experimental work was performed in the Environmental Molecular Science Laboratory, a DOE/BER national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, Washington. PNNL is operated for the DOE by Battelle under contract DE-AC05-76RLO-1830. References • Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, et al. (2013) Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest 123: 517-525. • Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. ProcNatlAcadSci U S A 102: 15545-15550. • Ideker T, Ozier O, Schwikowski B, Siegel AF (2002) Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18 Suppl 1: S233-240 • McDermott JE, Costa M, Janszen D, Singhal M, Tilton SC (2010) Separating the drivers from the driven: Integrative network and pathway approaches aid identification of disease biomarkers from high-throughput data. Dis Markers 28: 253-266 Caspase Integrin A9B1 PDGFRB RAC1 IL8-CXCR1 Alphasynuclein Genomically-defined subtypes [1] Genomically-defined subtypes [1] Module 1 (short survival) Topological Analysis of Association Networks Global phosphoproteomics Global proteomics Phosphoproteomics Proteomics Proteomics Phosphoproteomics Genomic Gene expression Clinical outcomes Genomic subtypes Association Networks Subtype analysis Activated in short survival * * Activated in long survival P = 0.007 * Comparisons WRI TCGA Generate an integrated co-expression/co-abundance network - Integrated transcriptomics, proteomics, and phosphoproteomics data - Statistical network inference across all samples - Hold out survival data and other genomic data Identifies active subnetworks[3] from co-abundance network - Searches for regions of network enriched in correlation with survival Assesses functional coherence of subnetwork modules to Infer drivers of cancer progression - Module members - Topologically important locations - Underlying genetic alterations Functional pathway analysis Module 2 (long survival) Network analysis Topological positions in the association network were assessed for statistical enrichment in known cancer drivers and pathogen targets. Asterisks indicate significance (p <0.001). This approach identifies genes/proteins that control information flow in the system and are therefore potential drivers of disease progression [4]. P = 0.005 Correlated with short survival Correlated with long survival Protein Phosphorylated protein mRNA What are the functional- and pathway-level correlates of survival in ovarian cancer? CONTACT: Jason McDermott Biological Sciences Division Pacific Northwest National Laboratory E-mail: jason.mcdermott@pnnl.gov Kaplan-Meier survival based on mutation, CNV, and mRNA expression for five gene signatures from network modules (http://www.cbioportal.org)