Download
1 / 36
360 likes | 649 Views
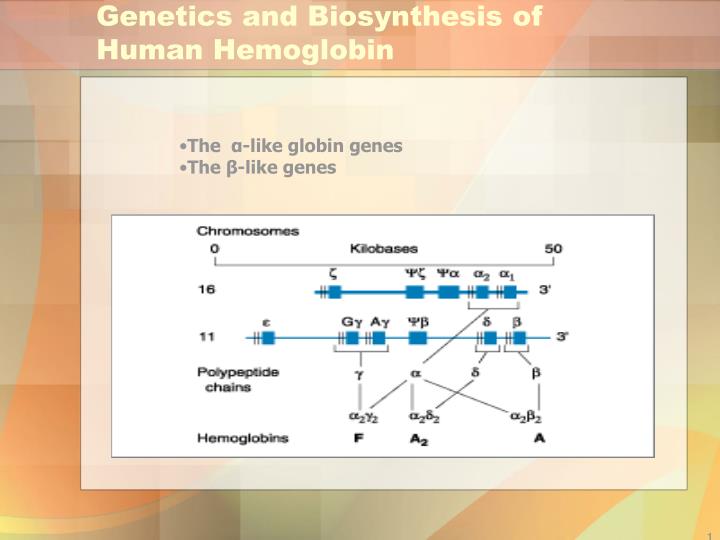
Genetics and Biosynthesis of Human Hemoglobin. The α -like globin genes The β -like genes. Hemoglobinopathies. HEMOGLOBIN STRUCTURE A pair of α -like chains 141 amino acids long and a pair of β -like chains 146 amino acids long.

E N D
Genetics and Biosynthesis of Human Hemoglobin • The α-like globin genes • The β-like genes
Hemoglobinopathies HEMOGLOBIN STRUCTURE A pair of α-like chains 141 amino acids long and a pair of β-like chains 146 amino acids long. The major adult hemoglobin is HbA (α2β2 ). HbF (α2γ2) predominates during most of gestation, and HbA2 (α2δ2) is a minor adult hemoglobin.
Hemoglobin Structure • Heme: a protoporphyrin IX ring complexed with a single iron atom in the ferrous state (Fe2+) • Every molecule of hemoglobin can thus transport up to four oxygen molecules. *
Hemoglobin Structure The hemoglobin tetramer is highly soluble, but individual globin chains are insoluble. Unpaired globin precipitates, forming inclusions (Heinz bodies) that damage the cell. Normal globin chain synthesis is balanced so that each newly synthesized α or non-α globin chain will have an available partner with which to pair to form hemoglobin.
Hemoglobin Developmental Biology • Red cells first appearing at about 6 weeks after conception contain the embyronic hemoglobins: • Hb Portland (ζ2γ2) • Hb Gower I (ζ2ε2) • Hb Gower II (α2ε2). • At 10 to 11 weeks, fetal hemoglobin (HbF; α2γ2) becomes predominant. The switch to nearly exclusive synthesis of adult hemoglobin (HbA; α2β2) occurs at about 38 weeks.
Hemoglobin Developmental Biology Fetuses and newborns therefore require α-globin but not β-globin for normal gestation.
Thalassemia Syndromes Mutations Transcription, processing of the mRNA precursor, translation, and posttranslational metabolism of the β-globin polypeptide chain. Deletions
Thalassemia Syndromes EPIDEMIOLOGY Thalassemias are the most common genetic disorders in the world, affecting nearly 200 million people worldwide.
Thalassemia Syndromes α-thalassemia trait (minor) occurs in 1 to 15% of persons of Mediterranean origin. β-Thalassemia has a 10 to 15% incidence in individuals from the Mediterranean and Southeast Asia.
β-Thalassemia Syndromes Clinical definitions β-thalassemia major (β0) β-thalassemia intermedia (β+) β-thalassemia minor
β-Thalassemia Major Profound anemia Erythropoietin release and compensatory erythroid hyperplasia Ineffective erythropoiesis Extramedullary erythropoietic tissue in the liver and spleen.
β-Thalassemia Major • Massive bone marrow expansion : characteristic "chipmunk" facies, thinning and pathologic fracture of long bones and vertebrae, and profound growth retardation.
β-Thalassemia Major Hemolytic anemia causes hepatosplenomegaly, leg ulcers, gallstones, and high-output congestive heart failure.
β-Thalassemia Major The shift of caloric resources to support erythropoiesis leads to: Inanition Susceptibility to infection, Endocrine dysfunction Death during the first decade of life.
β-Thalassemia Major Chronic transfusions: Iron overload, usually prove fatal by age 30. HbF persists to various degrees reducing the burden of α-globin inclusions.
β-Thalassemia Major DIAGNOSIS The diagnosis of β-thalassemia major is readily made during childhood on the basis of severe anemia accompanied by hepatosplenomegaly; profound microcytosis; a characteristic blood smear; and elevated levels of HbF, HbA2, or both.
Thalassemia SyndromesHb Electrophoresis HbA2 is frequently elevated in β-thalassemia trait and depressed in iron deficiency. HbF is elevated in HPFH, some β-thalassemia syndromes, and occasional periods of erythroid stress or marrow dysplasia.
β-Thalassemia Major Complete characterization: Amino acid sequencing Gene cloning and sequencing
β-Thalassemia Major MANAGEMENT Many patients require chronic hypertransfusion therapy designed to maintain a hematocrit of at least 27 to 30% so that erythropoiesis is suppressed.
β-Thalassemia Major MANAGEMENT Splenectomy is required if the annual transfusion requirement (volume of RBCs per kilogram body weight per year) increases by >50%. Vaccination with pneumococcal vaccine in anticipation of eventual splenectomy is advised.
β-Thalassemia Major MANAGEMENT Folic acid supplements may be useful. close monitoring for infection, leg ulcers, and biliary tract disease is needed.
β-Thalassemia Major MANAGEMENT Early endocrine evaluation is required for glucose intolerance, thyroid dysfunction, and delayed onset of puberty or secondary sexual characteristics. Many patients develop endocrine deficiencies as a result of iron overload.
β-Thalassemia Major MANAGEMENT Marrow transplantation from an HLA-identical sibling. Adequate iron chelation therapy after 100 U of PRBC (Desferoxamine, Deferasirox).
Thalassemia Syndromes β-Thalassemia minor (i.e., thalassemia trait) Profound microcytosis and hypochromia Target cells Minimal or mild anemia RBC number is higher than normal RDW is normal MCV is rarely >75 fL Elevated HbA2 (3.5 to 7.5%) Some forms are associated with normal HbA2 and/or elevated HbF. Genetic counseling and patient education are essential.
Thalassemia Syndromes β-thalassemia minor Misdiagnosis with I.D.anemia Iron usage is allowed if necessary
Alpha Thalassemia • Silent: αα/α- • Trait: αα/-- , α-/α- • Hb H disease: α-/-- • Hydrops fetalis: --/--
Thalassemia Intermedia Patients with β-thalassemia intermedia exhibit similar stigmata but can survive without chronic hypertransfusion. Management is particularly challenging because a number of factors can aggravate the anemia, including infection, onset of puberty, and development of splenomegaly and hypersplenism. Some patients may eventually benefit from splenectomy. The expanded erythron can cause excess absorption of dietary iron and hemosiderosis, even without transfusion.
Thalassemia Syndromes PREVENTION Antenatal diagnosis of thalassemia syndromes is now widely available. DNA diagnosis is based on PCR amplification of fetal DNA, obtained by amniocentesis or chorionic villus biopsy followed by hybridization to allele-specific oligonucleotides probes. The probes can be designed to detect simultaneously the subset of mutations that account for 95 to 99% of the a or b thalassemias that occur in a particular ethnic group.