Download
1 / 1
10 likes | 95 Views
Identification of Amino Acids that are Critical for Structural Stability and Functionality within the Heterodimerization (HD) Domain of Notch Proteins Marina Pell ón Consunji and Lucien Celine Montenegro Advisor: Dr. Didem Vardar-Ulu Wellesley College, Massachusetts.

E N D
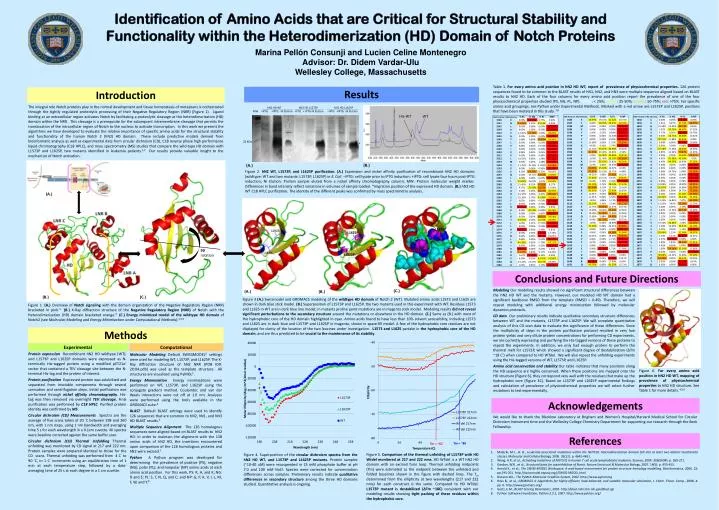
Identification of Amino Acids that are Critical for Structural Stability and Functionality within the Heterodimerization (HD) Domain of Notch Proteins Marina Pellón Consunji and Lucien Celine Montenegro Advisor: Dr. Didem Vardar-Ulu Wellesley College, Massachusetts Table 1. For every amino acid position in hN2 HD WT, report of prevalence of physicochemical properties. 126 protein sequences found to be common to the BLAST results of hN1, hN2, and hN3 were multiple sequence aligned based on BLAST results to hN2 HD. Each of the four columns for every amino acid position report the prevalence of one of the four physicochemical properties studied (PS, NG, PL, NP). White: < 25%; yellow: 25-50%; orange: 50-75%; red: >75%. For specific amino acid groupings, see Python under Experimental Methods. Marked with a red arrow are L1573P and L1625P, positions that have been mutated in this study. 7,8 Results Introduction The integral role Notch proteins play in the normal development and tissue homeostasis of metazoans is orchestrated through the tightly regulated proteolytic processing of their Negative Regulatory Region (NRR) (Figure 1). Ligand binding at an extracellular region activates Notch by facilitating a proteolytic cleavage at the heterodimerization (HD) domain within the NRR. This cleavage is a prerequisite for the subsequent intramembrane cleavage that permits the translocation of the intracellular region of Notch to the nucleus to activate transcription. In this work we present the algorithms we have developed to evaluate the relative importance of specific amino acids for the structural stability and functionality of the human Notch 2 (hN2) HD domain. These include predictive models derived from bioinformatic analysis as well as experimental data from circular dichroism (CD), C18 reverse phase high performance liquid chromatography (C18 HPLC), and mass spectrometry (MS) studies that compare the wild-type HD domain with L1573P and L1625P, two mutants identified in leukemia patients.1,2 Our results provide valuable insight to the mechanism of Notch activation. hN2 HD WT hN2 HD L1573P hN2 HD L1625P . MW –IPTG +IPTG Ni Elution -IPTG + IPTG Ni Elution –IPTG +IPTG Ni Elution WT His-WT 22 kDa * • q (B.) (A.) Figure 2. hN2 WT, L1573P, and L1625P purification. (A.) Expression and nickel affinity purification of recombinant hN2 HD domains (wildtype: WT and two mutants: L1573P, L1625P) in E. Coli. –IPTG: cell lysate prior to IPTG induction; +IPTG: cell lysate four hours post-IPTG induction; Ni Elution: Protein sample eluted from a nickel affinity chromatography column; MW: Protein molecular weight marker. Differences in band intensity reflect variations in volumes of sample loaded. *migration position of the expressed HD domain. (B.) hN2 HD WT C18 HPLC purification. The identity of the different peaks was confirmed by mass spectrometry analysis. (A.) L1625P L1625P 90° rotation L1573P L1573P Conclusions and Future Directions Modeling Our modeling results showed no significant structural differences between the hN2 HD WT and the mutants. However, our modeled HD WT domain had a significant backbone RMSD from the template (RMSD = 0.49). Therefore, we will repeat modeling with additional energy minimization followed by molecular dynamics protocols. CD data Our preliminary results indicate qualitative secondary structure differences between WT and the mutants, L1573P and L1625P. We will complete quantitative analysis of this CD scan data to evaluate the significance of these differences. Since the multiplicity of steps in the protein purification protocol resulted in very low protein yields and very dilute protein concentrations for performing CD experiments, we are currently expressing and purifying the His-tagged versions of these proteins to repeat the experiments. In addition, we only had enough protein to perform the thermal melt for L1573P, which showed a significant degree of destabilization (ΔTm ~18 C°) when compared to HD WTdel. We will also repeat the unfolding experiments using the His-tagged versions of WT, L1573P, and L1625P. Amino acid conservation and stability Our table indicates that many positions along the HD sequence are highly conserved. When these positions are mapped onto the HD structure (Figure 6), they correspond very well with the residues that make up the hydrophobic core (Figure 3C). Based on L1573P and L1625P experimental findings and calculation of prevalence of physicochemical properties we will select further mutations to test experimentally. (B.) (C.) Figure 3 (A.) Swissmodel and GROMACS modeling of the wildtype HD domain of Notch 2 (WT). Mutated amino acids L1573 and L1625 are shown in dark blue stick model. (B.) Superposition of L1573P and L1625P, the two mutants used in this experiment with WT. Residues L1573 and L1625 in WT are in dark blue line model; in mutants proline point mutations are in magenta stick model. Modeling results did not reveal significant perturbations to the secondary structure around the mutations or elsewhere in the HD domain. (C.) Same as (B.) with most of the hydrophobic core of the HD domain highlighted in cyan. Amino acids found to have less than 10% solvent accessibility, including L1573 and L1625 are in dark blue and L1573P and L1625P in magenta, shown in space-fill model. A few of the hydrophobic core residues are not displayed for clarity of the location of the two leucines under investigation. L1573 and L1625 partake in the hydrophobic core of the HD domain, and are thus predicted to be crucial to the maintenance of its stability. (C.) (A.) (B.) Figure 1. (A.) Overview of Notch signaling with the domain organization of the Negative Regulatory Region (NRR) bracketed in pink.3(B.) X-Ray diffraction structure of the Negative Regulatory Region (NRR) of Notch with the Heterodimerization (HD) domain bracketed orange.3 (C.) Energy minimized model of the wildtype HD domain of Notch2 (see Molecular Modeling and Energy Minimization under Computational Methods).4,5,6 Methods Experimental Computational Protein expression Recombinant hN2 HD wildtype (WT), and L1573P and L1625P domains were expressed as N-terminally His-tagged protein using a modified pET21a+ vector that contained a TEV cleavage site between the N-terminal His-tag and the protein of interest. Protein purification Expressed protein was solubilized and separated from insoluble components through several sonication and centrifugation steps. Initial purification was performed through nickel affinity chromatography. His-tag was then removed via overnight TEV cleavage. Final purification was performed by C18 HPLC. Purified protein identity was confirmed by MS. Circular dichroism (CD) Measurements Spectra are the average of five scans taken at 20 °C between 198 and 260 nm, with 1 nm steps, using 1 nm bandwidth and averaging time 5 s for each wavelength in a 0.1cm cuvette. All spectra were baseline corrected against the same buffer scan. Circular dichroism (CD) Thermal Unfolding Thermal unfolding was monitored by CD signal at 217 and 222 nm. Protein samples were prepared identical to those for the CD scans. Thermal unfolding was performed from 4 C° to 90 °C, in 1 C° increments using an equilibration time of 1 min at each temperature step, followed by a data-averaging time of 25 s at each degree in a 1 cm cuvette. Molecular Modeling Default SWISSMODEL4 settings were used for modeling WT, L1573P, and L1625P. The X-Ray diffraction structure of hN2 NRR (PDB ID#: 2OO4.pdb) was used as the template structure. All structures are visualized using PyMOL5. Energy Minimization Energy minimizations were performed on WT, L1573P, and L1625P using the conjugate gradient method. Coulombic and van der Waals interactions were cut off at 1.0 nm. Analyses were performed using the tools available in the GROMACS suite.6 BLAST Default BLAST settings were used to identify 126 sequences that are common to hN2, hN1, and hN3 HD BLAST results.7 Multiple Sequence Alignment The 126 homologous sequences were aligned based on BLAST results to hN2 HD. In order to maintain the alignment with the 138 amino acids of hN2 HD, the insertions encountered upon comparison of the 126 homologous proteins and hN2 were excised.7 Python A Python program was developed for determining the prevalence of positive (PS), negative (NG), polar (PL), and nonpolar (NP) amino acids at each amino acid position. For this work, PS: R, H, and K; NG: D and E; PL: S, T, N, Q, and C; and NP: G, P, A, V, I, L, M, F, W, and Y.8 Figure 6. For every amino acid position in hN2 HD WT, mapping of prevalence of physicochemical properties to hN2 HD structure. See Table 1 for more details. 4,5,6 Acknowledgements We would like to thank the Blacklow Laboratory at Brigham and Women’s Hospital/Harvard Medical School for Circular Dichroism Instrument time and the Wellesley College Chemistry Department for supporting our research through the Beck Fellowship. References Tm = ~80 Tm = ~62 • Malecki, M.J., et al., Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Molecular and Cellular Biology, 2006. 26(12): p. 4642-4651. • Weng, A.P., et al., Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science, 2004. 306(5694): p. 269-271. • Gordon, W.R., et al., Structural basis for autoinhibition of Notch. Nature Structural & Molecular Biology, 2007. 14(5): p. 455-455. • Arnold K., et al., The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics, 2006. 22: pp.195-201. http://swissmodel.expasy.org//SWISS-MODEL.html • DeLano W.L., The PyMOL Molecular Graphics System, 2002. http://www.pymol.org • Hess B., et al., GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation, J. Chem. Theor. Comp., 2008. 4: pp. 0. http://www.gromacs.org/ • Gertz, E.M., BLAST Scoring Parameters, 2005. http://blast.ncbi.nlm.nih.gov/Blast.cgi • Python Software Foundation, Python 2.5.1, 2007. http://www.python.org/ Figure 5. Comparison of the thermal unfolding of L1573P with HD Wtdel monitered at 217 and 222 nms. HD WTdel is a WT hN2 HD domain with an excised furin loop. Thermal unfolding midpoints (Tm) were estimated as the midpoint between the unfolded and folded baselines shown in the figure with dashed lines. The Tm determined from the ellipticity at two wavelengths (217 and 222 nms) for each construct is the same.Compared to HD WTdel, L1573P mutant is destabilized (ΔTm ~18C) consistent with our modeling results showing tight packing of these residues within the hydrophobic core. Figure 4. Superposition of the circular dichroism spectra from the hN2 HD WT, and L1573P and L1625P mutants. Protein samples (~10-40 uM) were resuspended in 25 mM phosphate buffer at pH 7.0 and 100 mM NaCl. Spectra were corrected for concentration differences across samples. Preliminary results indicate qualitative differences in secondary structure among the three HD domains studied. Quantitative analysis is ongoing.






