Download

1 / 69
690 likes | 1.16k Views
Molecular Orbitals several atoms. Any group of atoms: Molecules, Molecular ions, Fragments, Supermolecules. Describing molecular properties as a whole .

E N D
Molecular Orbitalsseveral atoms Any group of atoms: Molecules, Molecular ions, Fragments, Supermolecules
Describing molecular properties as a whole Molecular orbital theory is a method for determining molecular structure in which electrons are not assigned to individual bonds between atoms, but are treated as moving under the influence of the nuclei in the whole molecule.
The orbitalar approximation Molecular orbital is a function that describes the wave-like behavior of a single electron in a molecule. A polyelectronic wave-function is expressed in terms (as a product or a determinant) of MOs. The use of the term "orbital" was first used in English by Robert S. Mulliken in 1925 as the English translation of Schrödinger's use of the German word, 'Eigenfunktion'. Robert Sanderson Mulliken 1996-1986 Nobel 1966
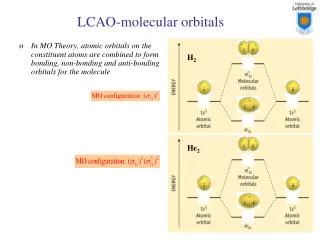
Generalizing theLCAO approach:A linear combination of atomic orbitals or LCAO It was introduced in 1929 by Lennard-Jones with the description of bonding in the diatomic molecules of the first main row of the periodic table, but had been used earlier by Pauling for H2+. Linus Carl Pauling 1901-1994 Nobel 1962 Sir John Lennard-Jones 1894-1954
Any calculation starts by inputs and provides outputs Input Definition of the system Total number of electrons Choice of state (ground state or excited state) Nature of atoms involved (Z) Atomic Orbitals Geometry to define Potential Output Energy, Molecular Orbitals Electronic density Spin, Magnetism Search for minimization of energy The calculated geometry is the minimum found by optimization
Structure and Reactivity How the energy of a system respond to a structural variation? Changing distance or angle.
Symmetry • To describe a molecule is describing • Its geometry and its symmetry • Its energy • MOs are eigenfunctions of the symmetry operator and of H • For H2, we have use symmetries without solving the Schrodinger equation, • For a larger system, symmetry helps simplifying or analyzing. • One common symmetry is the plane for planar molecules; this causes sand p separation. • To be considered when some parameter varies (structure modification, structure optimization, reactivity) symmetry has to be preserved during the variation
Valence orbitalssize of the orbitals Usually, the atomic orbitals that participate to the MOs are valence orbitals. These are the valence orbitals of the neutral atom. For a cation in its highest oxidation state, this might be the unoccupied shell whereas the size of the cation is given by the outermost occupied shell See the case of Li+
Valence orbitalssize of the orbitals, the Li+ example Size of the cation Li+ according to Slater? The last occupied shell is the 1s Z=(3-0.31) <r> = 1.5/2.69 a0 = 0.558 a0 = 0.295 Ả This is small (The Pauling ionic radius is also small, 0.60 Ả) The valence orbital 2s is the one involved in forming bonds Z=(3-2*0.85) <r> = 5/1.3 a0 = 3.846 a0 = 2.034 Ả Atomic radius (empirical): 1.45 Ả Atomic radius (calculated): 1.67 Ả Covalent radius (2008 values): 1.28 Ả Covalent radius (empirical): 1.34 Ả van der Waals radius: 1.82 Ả The covalent radius for the 2s orbital is such that the orbitals overlap; it is less than the <r> value
Method to build M.O.s • Determine the symmetry elements of the molecule • Make the list of the functions involved (valence atomic orbitals) • Classify them according to symmetry (build symmetry orbitals if necessary by mixing in a combination the set of orbitals related by symmetry) • Combine orbitals of the same symmetry (whose overlap is significant and whose energy levels differ by less than 10 eV).
Valence orbitals of H2O Symmetry relative to the molecular plane, sigma-pi separation
Valence orbitals of H2O Symmetry relative to xOz, necessitates building symmetry orbitals Build them ! What are their energy levels?
Valence orbitals of H2O Redundancy: Three groups; A2 is not present
Symmetry orbitals The H-H distance is 2*sin(105/2)*1.09 Ả = 1.73 Ả This is long relative to 0.74 Ả ! The energy level of the symmetry orbital is the atomic level for H, -13.6 eV (1s1+1s2) (1s1-1s2)
Values from Extended Hückel 1962(-eV) H He 1s 13.6 24.25 Li Be B C N O F 2s 5.4 10 15.2 21.4 26 32.3 40 2p 3.5 6 8.5 11.4 13.4 14.8 18.1 Na Mg Al Si P S Cl 3s 5.1 9 12.3 17.3 18.6 20 30 3p 3. 4.5 6.5 9.2 14 13.3 15 Roald Hoffmann Nobel 1981 William Nunn Lipscomb, Jr. American 1919- Nobel 1976
1 Valence orbital of H2O Symmetry B1 The atomic orbital 2pxO is a lone in its group, It is a molecular orbital It has p symmetry. E2p(O)= -14.8 eV
2 Valence orbitals of H2O Symmetry B2 E2p(O)= -14.8 eV and E1S(H) = -13.6 eV
3 Valence orbitals of H2O Symmetry A1 E2s(O)= -32.3 eV E2p(O)= -14.8 eV And E1S(H) = -13.6 eV E2s is low in energy an d does not mix
6 AO → 6 MOs 3 A1 → 3 A1 2B2 → 2B2 1 B1 → 1 B1 What is the ordering of the levels?
Overlap involved in B2 Overlap involved in A1 At 45° same overlap angle HOH/2 = 105/2 = 52°5 > 45° Splitting in B2 > Splitting in A1
2B2 6 AO → 6 MOs 3 A1 → 3 A1 2B2 → 2B2 1 B1 → 1 B1 4A1 In the Lewis formula, two electron pairs, two bonds The electron pairs are 2s and 2p Spectroscopy shows different electron levels The bonds are the result of two contributing MO A1 and B2 3A1 1B2 Orbital numbering: 1A1 for the core? 2A1
MOs Hybridization sp3 agreement with VSEPR and Total density sp2 closer to MOs and spectroscopy: two different pairs
Typical Bond Angles in AH2Moleculesmolecule electronic configuration H-A-H bond angle 1a1= 1sg ; 1b2= 1su
C 3 Valence orbitals of CH2 Symmetry A1 E2s(C)= -21.4 eV E2p(C)= -11.4 eV And E1S(H) = -13.6 eV E2sC – E1sH < 10 eV Hybrid orbitals Not atomic eigenfunctions But part of the MOs eigenfunctions for the molecule 2s-2pZ 2s+2pZ A 2A1 is bonding 3A1 is non-bonding 4A1 is antibonding
C 3 Valence orbitals of CH2 Symmetry A1 E2s(C)= -21.4 eV E2p(C)= -11.4 eV And E1S(H) = -13.6 eV E2sC – E1sH < 10 eV A Among the 3 A1 valence MO, the lowest one is bonding and the middle one non-bonding; this differs from H2O!
hydridization Justification: • s and pZ both are of same symmetry and appear in the same linear combinations (MOs); hybridization allows anticipating. • hybridization aims combining AOs, each hybrid maximizing the interaction with a partner (here the symmetry orbital on Hs) and minimizing those with others. The idea is that we can thus separate the interactions with different partners. If correct, this is useful and allows transferability (replacing a substituent by another one involving the same hybrid). • Mathematically a set of hybrids is equivalent to a set of canonic orbitals Failure: • It is not possible to rigorously separate the interactions with different partners. Hybrids interact with each other. • Existing symmetries reappear if interactions between hybrids is included.
Methane The MO orbital description conflicts with sp3 hybridization
sp3 hybridization <tiItj>= δij <tiIHItj>= (E2s-E2p)/4 Eti = <tiIHIti> = (E2s+3E2p)/4
sp3 hybridization With b =(E2s-E2p)/4 = 0 E2p 1(E2s-E2p)/4 (E2s+3E2p)/4 3 (E2s-E2p)/4 E2s
sp3 hybrids along C3 ¼ of 2s 3/4 of 2p
Interest of hybridization • Pictorial • Anticipating AO combinations within MOs • Global density • Transferable: Analysis through substituents • Writing VB structures What is bad with hybridization? • Not describing symmetry • Not good for spectroscopy • Lack of orthogonality between hybrid orbitals belonging to the same center (tails of localized orbitals)
hybridization Mixing 2s and 2p: requires degeneracy to maintain eigenfunctions of AOs. Otherwise, the hybrid orbital is an average value for the atom, not an exact solution. This makes sense when ligands impose directionality: guess of the mixing occurring in OMs.
Angular dependence • Expression along C2 (z is the main axis) • t = a s + √(1-a2) [pzcosq ± px sinq] • t = a’s + √(1-a’2) [pzcosq’ ± py sinq’] • 2a2+2a’2=1 • √(1-a2) sinq = 1/√2 • √(1-a’2) sinq’ = 1/√2 • = 105.5/2 (<109°28) = 52.75° → a = 0.459 a’= √(1/2-a2) = 0.548 • sinq’ = (1/√2)/(1/ √(1-a’2)=0.8453 • q’ = 57°33 = 114.7/2 > 109°28 2q : 2q’ :
Angular dependence a2+a’2= ½ √(1-a2) sinq = 1/√2 √(1-a’2) sinq’ = 1/√2 (1-a2) sinq2 = 1/2 =(1-a’2) sinq’ 2 1-1/2sinq2 = a2 1-1/2sinq’2 = a’2 1-1/2sinq2 +1-1/2sinq’2 = a2 + a’2 2 = 1/2sinq2 +1/2sinq’2 +½ 3 = 1/sinq2 +1/sinq’2 When qdecreases q’increases! 2q : 2q’ :
Weight of s orbital 2q = 105°5 water a is smaller than ½ a’ is larger than ½ The weight of s is larger in the lone pairs. The s level is lower in energy than the p level. It has to participate to the stabilization of lone pairs. L-, CH3+ 90° pure p sp3 ligand 109°28’ ¼ s ¾ p CH3-,L+ >109° 28’ >1/4 s
For the reconstructed Silicon(100) surface, the s character is higher for the dangling bonds of the outmost atoms. - +
The orbitals of H3 (equilateral triangle in the xy plane) Are symmetry orbitals of NH3 and CH3 matching p orbitals H3 1/√2 -1/√2 -b 2px 2py 2b 2pz 1/√3
The orbitals of H3 (equilateral triangle in the xy plane) are symmetry orbitals of NH3 and CH3 matching p orbitals H3 1/√2 -1/√2 -b 2px 2py 2b 2pz 1/√3
Pseudo symmetry: for H3 the OM are degenerate. They remain degenerate interacting with the 2p orbitals (E) H3 2/3 = 1/√2 + c2 1/√2 -1/√2 -b 2px 2py √(2/3) Normalization 1 = (2/√3)2 +2 c2
The orbitals of H3 (equilateral triangle in the xy plane) are symmetry orbitals of NH3 and CH3 matching p orbitals H3 1/√2 -1/√2 -1/√6 -1/√6 -b 2px 2py √(2/3) 2b 2pz 1/√3
NH3 s system Lone pair Bonding