Download

1 / 20
200 likes | 379 Views
Computational Studies of Tryptophanyl-tRNA Synthetase: Activation of ATP by Induced-Fit. Kapustina, M. and Carter, C.W. (2006) J. Mol. Biol. 362:1159-1180. TrpRS - type I tRNA synthetase, 326 aa ( B. stearotherm. ) domains: RS – Rossman fold, dinucleotide binding domain

E N D
Computational Studies of Tryptophanyl-tRNA Synthetase: Activation of ATP by Induced-Fit Kapustina, M. and Carter, C.W. (2006) J. Mol. Biol. 362:1159-1180.
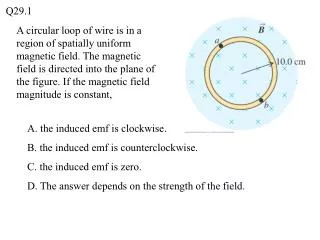
TrpRS - type I tRNA synthetase, 326 aa (B. stearotherm.) • domains: • RS – Rossman fold, dinucleotide binding domain • ABD – anti-codon binding domain • crystal structures • open: 1MAW (ATP), 1MB2 (Trp) • preTS: 1M83, 1MAU (ATP+Trp+Mg2+) • closed: 1I6L • conformational changes (induced fit) • small-scale: KMSKS catalytic loop (107-120) • large-scale: domain rotation between RS and ABD • Motivating questions: stability vs. ATP affinity paradox • open-form: Kd=0.4 mM ATP, 177 Å2 exposed surface area • preTS-form: Kd=~8 mM ATP, 23 Å2 exposed surface area • why does open-form bind ATP tighter, despite the fact that preTS makes more protein-ligand interactions and is less solvent accessible? • how is induced-fit triggered? how is preTS activated?
Computational Studies –Molecular Dynamics Simulations open, unliganded • advantages and disadvantages • SIGMA - Jan Hermans, UNC (based on CHARMM?) • time step: 2 fs • trajectories: up to 5000 ps (5 ns) • “validation”: compare mean positional RMSD to crystal B-factors • simulation under-estimates thermal motions • but there is relative correlation closed, liganded
Observations – MD simulation of open form • 4 cases: unliganded, Trp, ATP, ATP+TRP • all simulations were stable (no major changes) • loop is flexible: accounts for 40% of RMSD • open: <B107-120>=107.9 • open+Trp: <B107-120>=87.5 • open+ATP: <B107-120>=80.3 • opposite effects of ligand-binding on stability • Trp binds RF, reduces fluctuations in ABD • ATP binds ABD, increases fluctuations in RF
open, unliganded green: 150 ps magenta: 1200 ps wheat: 4500 ps blue: monomer magenta: monomer without loop 107-120
preTS – unstable without (both) ligands, reverts toward open-form • unliganded and Trp-only • rearrangement over 1-2 ns • ATP+Trp stabilizes preTS structure • preTS+Trp+ATP – progresses toward “closed” form (like with Trp-AMP product) • closed-form: stable, even without ligands
Characterizing domain rotations: a, g • a: hinge angle (bend), range: 10 deg • g: twist (rotation), range 9 deg Table 3. Average hinge and twist rotation angles
Interactions - 4 key lysines • acceptor loop: K109, K111 • KMSKS loop: K192, K195 • simulations of open form liganded with Trp+ATP show K109 in loop moves 14A to contact O in ATP triphosphate • however, K111 contacts triphosphate in preTS; probably exchange coordination • mutation K111A leads to rapid loss of twist in preTS – probably essential in assembly of induced fit • Trp approaches ATP; may cause ordering of 107-120 loop Trp Open form ATP
open form (1MAW), side view open form (1MAW), top view preTS form (1MAU), side view preTS form (1MAU), top view
preTS => product form • KSKMS loop moves 1.3A in product crystal structure, and 2.5A in preTS trajectories • probably separation of PPi
Effect of Mg2+ • does not affect domain rotation of fully-liganded preTS • no direct contacts with protein side-chains • Mg2+ coordinates triphosphate tail • 5-coordinate sites filled, 3 by O’s, 2 by waters • unusually long bond distances: 2.54A vs. 1.85A avg • preTS+ATP+Mg: • maintains a hinge, but g untwists like product state • preTS+Trp+ATP+Mg: • catalytic loop pops open after 2500 ps • without Mg, triphosphate uncoils, domains untwist (by 5-7 degrees)
unrestrained simulations: • Mg moves closer to triphosphate O’s • distrupts K111 and K192 interactions, causing loop excursions • add harmonic restraints • quadratic: E = ... + wSi(dist(Mg,Oi)-2.54)2 • use potential of mean force (PMF) to estimate force necessary to counter tendency to move Mg • try different force constants w till achieve balance • prevent 0.7A displacement • about 5% of strength of Coulombic interaction • with restraints, preTS simulations remain stable • domains do not untwist; lysines stay in contact
Mg and Lys192 “share” attraction to triphosphate; holds ABD in high-energy twist
Coupling of Mg:ATP:Lys192 interaction to domain rotations • restrain centers-of-mass with 500 kcal/mol.A2 to prevent hinge opening and ABD untwisting • Lys192 and Lys111 stay in contact with ATP O’s
Model of allosteric behavior • KNF (yes) – induced fit, tertiary changes propagate • MWC (no) – symmetry effect, quartenary coupling • preTS is stable only with ligands (ATP+Mg) • increased interactions of ATP with active site compensate for strain of domain-twisting • supplies “energy” for catalysis (?) • note: simulations done with monomer • negative cooperativity of dimer • also supports KNF (only one consistent with induced-fit)
Summary of Trp-tRNA synthetase binding and activation • formation of preTS by induced-fit • ATP binds, causes domain rotation, brings K109 (RF) and K192 (ABD) together • Trp binds, causes ordering of acceptor loop • Trp brought in contact with ATP • in preTS • K109 replaced by K111 • Mg binds triphosphate tail • Mg helps hold K192 and K111 in place (near triphosphate) • high Mg-O distances reflect strain in twisted state • catalysis • domains untwist (partially), but do not open up (hinge angle) • PPi moves with KSMKS loop as it opens up • Mg stabilizes transition state (AMP-1) for transfer to Trp (acylation)