Download
1 / 1
10 likes | 109 Views
HUGO: Hierarchical mUlti -reference Genome cOmpression tool for aligned short reads . Pinghao Li, 1 Xiaoqian Jiang, 2 Shuang Wang, 2 Jihoon Kim, 2 Hongkai Xiong, 1 and Lucila Ohno-Machado 2. 1 EE Department, Shanghai Jiaotong University, Shanghai, China

E N D
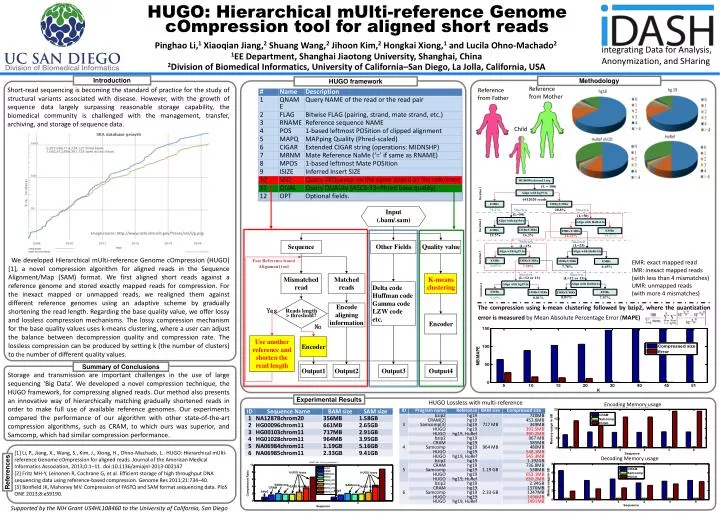
HUGO: Hierarchical mUlti-reference Genome cOmpression tool for aligned short reads Pinghao Li,1 Xiaoqian Jiang,2 Shuang Wang,2Jihoon Kim,2Hongkai Xiong,1 and Lucila Ohno-Machado2 1EE Department, Shanghai Jiaotong University, Shanghai, China 2Division of Biomedical Informatics, University of California–San Diego, La Jolla, California, USA Introduction Methodology HUGO framework Reference from Mother Short-read sequencing is becoming the standard of practice for the study of structural variants associated with disease. However, with the growth of sequence data largely surpassing reasonable storage capability, the biomedical community is challenged with the management, transfer, archiving, and storage of sequence data. Reference from Father Child Image source: http://www.ncbi.nlm.nih.gov/Traces/sra/i/g.png We developed Hierarchical mUlti-reference Genome cOmpression (HUGO) [1], a novel compression algorithm for aligned reads in the Sequence Alignment/Map (SAM) format. We first aligned short reads against a reference genome and stored exactly mapped reads for compression. For the inexact mapped or unmapped reads, we realigned them against different reference genomes using an adaptive scheme by gradually shortening the read length. Regarding the base quality value, we offer lossy and lossless compression mechanisms. The lossy compression mechanism for the base quality values uses k-means clustering, where a user can adjust the balance between decompression quality and compression rate. The lossless compression can be produced by setting k (the number of clusters) to the number of different quality values. EMR: exact mapped read IMR: inexact mapped reads (with less than 4 mismatches) UMR: unmapped reads (with more 4 mismatches) The compression using k-mean clustering followed by bzip2, where the quantization error is measured by Mean Absolute Percentage Error (MAPE) Summary of Conclusions Storage and transmission are important challenges in the use of large sequencing ‘Big Data’. We developed a novel compression technique, the HUGO framework, for compressing aligned reads. Our method also presents an innovative way of hierarchically matching gradually shortened reads in order to make full use of available reference genomes. Our experiments compared the performance of our algorithm with other state-of-the-art compression algorithms, such as CRAM, to which ours was superior, and Samcomp, which had similar compression performance. Experimental Results HUGO Lossless with multi-reference Encoding Memory usage [1] Li, P., Jiang, X., Wang, S., Kim, J., Xiong, H., Ohno-Machado, L.. HUGO: Hierarchical mUlti-reference Genome cOmpression for aligned reads. Journal of the American Medical Informatics Association, 2013;0:1–11. doi:10.1136/amiajnl-2013-002147 [2] Fritz MH-Y, Leinonen R, Cochrane G, et al. Efficient storage of high throughput DNA sequencing data using reference-based compression. Genome Res 2011;21:734–40. [3] Bonfield JK, Mahoney MV. Compression of FASTQ and SAM format sequencing data. PloS ONE 2013;8:e59190. Decoding Memory usage References