Download

1 / 42
420 likes | 538 Views
CONGESTIVE HEART FAILURE DEFINITION 1920's - " organ physiology " paradigm - interplay between the abnormal heart and the circulation. The focus on circulatory abnormalities 1960's - " cell biochemistry " paradigm - depressed contractility and impaired relaxation

E N D
CONGESTIVE HEART FAILURE DEFINITION 1920's - "organ physiology" paradigm - interplay between the abnormal heart and the circulation. The focus on circulatory abnormalities 1960's - "cell biochemistry" paradigm - depressed contractility and impaired relaxation 1980's - "gene expression" paradigm - molecular alterations in the myocardial cells Heart failure is a clinical syndrome in which impaired cardiac pumping decreasesejection and impedes venous return. These haemodynamic abnormalities are generally complicated by depressed myocardial contractility and relaxation, which reflect biochemical and biophysical disorders in the myocardial cells. This latter, in turn, are due to partly to molecular abnormalities that not only impair the heart'sperformance, but also acceletrate the deterioration of the myocardium and hastens myocardial cell death. PATHOPHYSIOLOGY OF HEART FAILURE Heart failure can develop: a.) Acutely • resulting from acute myocardial infarction • secondary to an infectious or infiltrating process (virus, bacterial, rotozoal) b.) Chronically (over months or years) as the end-stage of different heart diseases. This low output failure can result from: a.) decrease in myocardial contractile reserve, due to: • myocardial infarction • cardiomyopathy • increased afterload (eg. hypertension) b.) valvular disease (eg. aortic stenosis or mitral regurgitation) c.) prolonged rhythm disturbances (eg. ventricular tachycardia)
The primary signs and symptoms of all types of CHF include: • tachycardia • decreased exercise tolerance • shortness of breath • peripheral and pulmonary edema • cardiomegaly Cardinal feature: cardiac output (CO falls short of what is required for normal tissue perfusion Reason: decrease in cardiac contractility Low output failure: responds to positive inotropic drugs High output failure: the demands of the body are so great that even increased CO is insufficient (eg. hyperthyreodism, beri-beri, anaemia, arteriovenous shunt). Respond poorly to positive inotropic drugs. Haemodynamics of heart failure Forward or inotropic failure - reduced ejection into the aorta and pulmonary artery Backward or lusitropic failure - inadequate emptying of the venous reservoirs
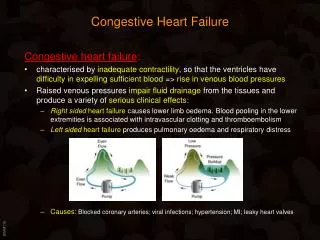
SITE OF FAILURE BACKWARD FAILURE FORWARD FAILURE Right heart failure Increased systemic Reduced ejection into venous pressure pulmonary artery Left heart failure Increased pulmonary Reduced ejection into the a venous pressure aorta Because blood flows in a circle, none of these occurs in pure form. In "Forward failure" - when the ventricle empties poorly, the filling is reduced In "Backward failure" - when filling is reduced, the stroke volume is reduced In right ventricular failure: the increase in systemic venous pressure and decreased ejection of blood into the pulmonary artery reduce the output of the left ventricle In left ventricular failure: increased pulmonary venous pressure impedes the blood out of the lungs increases pulmonary capillary pressure. This is transmitted across the pulmonary circulation and results in increased pulmonary arterial pressure which can impair right ventricular ejection.
Signs and symptoms of heart failure The clinical picture in heart failure consists of: • signs (the objective manifestations of depressed cardiac performance), • symptoms (abnormalities perceived by the patient). Left heart failure: is a "forward failure" - reduced ejection, "backward failure" - rise in pulmonary capillary pressure. Systemic reflex activation: vasoconstriction increased blood pressure (to maintain perfusion of vital organs; ie. heart, brain) Despite the increased sympathetic tone perfusion decreases • to the skeletal muscle: fatigue and skeletal muscle myopathy • to the kidneys: oliguria, sodium and water retention • to the tissues: cyanosis Because of the backward failure pulmonary congestion impaired respiration • Dyspnea (difficulty breathing) due to arterial hypoxia, and decreased lung compliance (excess fluid transudates from the pulmonary capillaries) depth and rate of breathing increase). Typical in supine position. Mild HF: dyspnea occurs only during heavy exercise Severe HF: dyspnea is present at rest (bubbling noises during respiration) End-stage HF: fluid fills the bronchial system; pulmonary edema Right heart failure: is a backward failure rather than a forward failure. • The central venous pressure is over the maximum. • Edema (liver, kidneys, spleen, GIT, skin, genitalies) • Cyanosis
Cardiomegaly - one of the major signs of heart failure. • Initially due to the operation of the Frank-Starling relationship • Remodelling of the ventricular wall is a complex process Pressure overload (hypertension, aortic stenosis `inward' hypertrophy, reduced ventricular cavity (concentric hypertropy) Volume overload (aortic regurgitation) the ventricle dilates (eccentrichypertrophy) Grading of severity of heart failure according to the New York Heart Association (NYHA) NYHA I. Signs of heart failure appears only heavy exertion and disappear after its discontinuation NYHA II. On normal workload signs of CHF appears in the evening but disappear after night rest NYHA III. At rest minimal signs of CHF causing no complains but marked signs of CHF on walking, which are not fully relieved by night rest. NYHA IV. Signs of CHF even at bed rest.
Compensatory mechanisms in heart failure Extrinsic reflex mechanisms for compensation: Sympathetic nervous system (SNS) Renin-angiotensin-aldosterone hormonal response. Increased sympathetic outflow: • tachycardia, • increased contractility, • increased vascular tone (venous tone) • increased ventricular filling pressure • dilatation of the heart • increased fiber stretch Increased aldosterone secretion: • sodium and water retention • increased blood volume • edema.
Intrinsic compensatory mechanism: myocardial hypertrophy • Increased muscle mass (to maintain cardiac performance) • Hypoxic myocardium • Decreased oxygen supply to the myocardium 0 0 5 5 10 10 15 15 20 20 25 25 2 1 normal Developed tension Velocity of contraction normal failing failing PCWP (Hgmm) LOAD Depressed contractility in heart failure reflected either as a reduced peak tension development (1) or depressed force-velocity curve (2)
Pathophysiology of cardiac performance Cardiac performance depends on at least 4 primary function: A: PRELOAD (LVEDP, LVEDV, reflected as central venous pressure) Preload refers to the diatolic loading conditions of the heart; - Left ventricle: left atrial pressure (or pulmonary capillary wedge pressure = PCWP) - Right ventricle: right atrial pressure for the right ventricle. These are the "filling pressures" Left ventricular function curve: (SV or SW against the filling pressure) The ascending limb (bellow 15 mmHg) represents the classic Frank-Starling relation. Beyond approximately 15 mmHg, there is a plateau of performance. Preload greater than 20-25 mmHg result in pulmonary congestion.
The Frank-Starling low of the heart describes the property of cardiac muscle to increase its contractility as the length of the myocardial fiber (stretch) is increased. To accomplish this increase in stretch, more blood must be returned to the heart by: a.) Increased sympathetic tone causing vasoconstriction decreased venous blood storage (pooling) increased end-diastolic volume (or filling pressure) and CO. b.) Redistribution of blood from viscera to heart c.) Fluid or sodium retention due to decreased renal perfusion, and renin-angiotensin- aldosterone activation. This increases volume of blood returned to the heart and also may cause edema. REDUCTION OF PRELOAD: DIURETICS B: AFTERLOAD: is the resistance against which the heart must pump blood. Systemic vascular resistance is frequently increased in CHF (increased sympathetic outflow and circulating catecholamines). This may speed failure. REDUCTION OF AFTERLOAD: ARTERIAL VASODILATORS C: CONTRACTILITY: is the vigor of contraction of heart muscle. In CHF the primarily defect: reduction in the intrinsic contractility (dP/dt) INCREASE IN CONTRACTILITY: POSITIVE INOTROPIC DRUGS. D: HEART RATE: is the major determinant of cardiac output (CO) - When CO decreases HR increases (beta-adrenoreceptor activation) -Consequences: - diastole shortens - myocardial perfusion worsens - hypoxia
CARDIAC HYPERTROPHY The most important intrinsic compensatory mechanisms. Complex biochemnical biophysical mechanisms in the background. a.) Energetics in the failing heart Imbalance betwen energy production and energy utilization. Cause: is the overload itself Result: "state of energy starvation" (increased energy utilization and decreased high energy phosphate production). b.) Structural changes in the chronically overloaded heart Failing heart is not equal with a normal enlarged heart (sportmen’s heart) Architectural changes: Pressure overload - the walls of the heart thicken Volume overload - dilated heart Chronic heart failure: hyperthrophied heart: • myocyte necrosis (fibroblast poliferation) • muscle is replaced by connective tissue • the heart begins to dilate • wall tension incerases • propensity for arrhythmias c.) Altered blood supply • Imbalance between the capillary density and muscle mass (increased intercapillary distance) • Decreased coronary reserve (underperfused subendocardium) • Subendocardial necrosis
d.) Altered proportion of mitochondria and myofibrils • Imbalance between myofibrils and mitochondria (more energy-consuming myofibrils must be supplied with ATP by relatively fewer mitochondria) • Exacerbated energy starvation NOTE: a.) Many of these sings and symptoms of CHF are a direct result of these compensatory mechanisms. b.) Despite all attempts to compensate, the cardiac function deteriorate VITIOUS CYCLE occur, and CO cannot be maintained without medical intervention Arrhythmogenic mechanisms in the hyperthropied heart • Enlargement and fibrosis of the atria and ventricles: increased susceptibility to arrhythmias (slow conduction reentrant arrhyhtmias) • Increased calcium accumulation in the cells initiate triggered activity. • Lowered resting potential (sodium pump inhibition) slow conduction. • Acidosis slowing of conduction SUDDEN CARDIAC DEATH Altered gene expression in the chronically overloaded, failing heart Gives the answer; Why the prognosis is so poor in this patients? • Appearance of abnormal proteins (accelerated potein synthesis) • Abnormalities in gene expression (accelerate the deterioration) The detrimental consequences of hypertrophy seems to represent a `price' that the overload heart must pay in order to accelerate protein sysnthesis.
The heart's response to overload can be divided in three phases that have different functional and prognostic implications. a.) First short-term stage of acute heart failure Clinical: left heart failure, pulmonary congestion Pathological: dilatation of the left ventricle Histological: swelling and separation of myofibrils Biochemical: glicogene and ATP levels decreased, lactate slightly increased b.) Second long-term stage of compensatory hyperfunction Clinical: relief of symptoms Pathological: hypertrophy Histological: increased size of cardiac fibers, minimal fibrosis Biochemical: glycogen, ATP normal, lactate increased. Miofibrillar mass increased relative to that of the mitochondrial mass. 3.) Third long-term stage of progressive exhaustion, cell death and fibrosis Clinical: reappearance of heart failure Pathological: fibrous replacement of muscular tissue Histological: connective tissue, fatty dystrophy Biochemical: as in second stage, exept decline in protein systhesis and marked decline in DNA levels.
NEUROHUMORAL AND RENAL MECHANISMS IN HEART FAILURE Neurohumoral systems may play both a detrimental and protective role in the pathogenesis of CHF. Compensatory mechanisms activated in CHF: vasoconstrictive and antinatriuretic vasodilatory and natriuretic The biologic activities of these systems are antagonistic. Vasoconstrictive-antinatriureticVasodilatory-natriuretic Renin-angiotensin-aldosterone system Atrial natriuretic factor (ANF) Sympathetic nervous system (SNS) Prostaglandins Vasopressin Dopamine Thromboxane Kallikrein, kinins Endothelin EDRF 1. Vasoconstrictive-antinatriuretic system a.) Sympathetic nervous system (SNS) Increased activity of SNS occurs early in the course of CHF and contributes to clinical deterioration and mortality in HF (1) Adrenergic receptor function Increased sympathetic activity receptor down-regulation ACE inhibitors (eg. Captopril) resensitization (reduced NA release) Evidence for: decreased level of Gs and increased level of Gi Future tool? (2) Baroreceptor or baroreflex abnormalities in heart failure Baroreflex control of the heart is impaired (structural changes in baroreceptors or arterial wall).
(3) Sympathetic stimulation is involved in: • LV remodelling • loss of myocardial cells • gene expression Increased sympathetic drive induces: a.) myocardial hypertrophy and fibroblast hyperplasia through stimulation of 1 and receptors b.) accelerated myocardial cell lost - apoptosis c.) NA may induce apoptosis through receptor activation d.) changes in myocardial gene expression which results in progressive worsening in contractility (downregulation of the adult type, high ATP-ase activity -myosin heavy chain isoform and upregulation of the fetal type, low ATP-ase activity -myosin heavy chain isoform) b.) Renin-angiotensin system Angiotensin II (AGII) is the key element with multiple biologic activity AG II influences cardiac metabolism, involved in the development of ventricular hypertrophy through its growth promoting effects. Interaction with other neurohumoral systems (enhances NA synthesis, reduces NA reuptake, facilitates NA release from nerve endings). Antagonistic with the ANF system
Plasma RAS maintains circulatory homeostasis during acute and subacute alterations in cardiac output Tissue RAS contributes to maintain homeostasis during impairment of CO 1. Activation of the RAS Occurs in response to reduction in cardiac output. Arterial constriction SABP and DABP Catecholamine release Angiotensin II. aldosteron secretion (adrenal cortex) • sodium retention • water retention • potassium excretion Increased blood pressure • water intake • vasopressin secretion • adrenocorticotropic hormon AGII binding to AT1 receptor leads to cardiac remodelling which includes: • upregulation of many early active cardiac myocyte genes • induction of late markers of cardiac hypertrophy (a-actin) and growth factors such as TGF • shift to fetal type myocardium AGII binding to AT2 receptor may oppose the effect of AGII on AT1 receptors
c.) Vasopressin Crutial role in hyponatremia in CHF (V2 linked hydro-osmotic effect) In small concentrations vasodilator (EDRF release?) 2. Vasodilatory-natriuretic systems a.) Atrial natriuretic factor (ANF) Maintains renal haemodynamic function, attenuates the RAS system ANF levels are elevated in CHF (indicator of developing dysfunction) In CHF relative ANF deficiency Therapeutical value: ANF replacement ? b.) Prostaglandins Endogenous vasodilator PGs (PGI2) opposing the effect of the vasoconstrictive endogenous agents c.) Dopamine Therapeutic agent in CHF d.) Endothelium derived vasoactive agents Both vasodilator (EDRF -- NO ?) and vasoconstrictor (endothelin) Endothelin - renal and systemic vasoconstrictor, activates RES
CELLULAR MECHANISMS IN HEART FAILURE 1. Role of calcium levels • Calcium regulates - contraction/and relaxation. • Intracellular calcium is modulated by: cAMP and IP3 "second messenger" systems • Extrusion of calcium is regulated by: calcium pump (CaATP-ase) and calcium- sodium exchange • In CHF abnormal calcium handling is apparent 2. cAMP It is an important "second messenger" modulates calcium handling • activates protein kinases (Ca2+ entry) • regulates calcium sequestration into the SR • regulates myofilament responsiveness to calcium In CHF reduced cAMP levels are apparent. 3. Sarcolemmal receptors and mechanisms • Homologous receptor downregulation cAMP dependent protein kinase phosphorilates the receptors desensitization • In CHF the SR cacium uptake function is altered 4. Altered myocardial responsiveness to calcium • In CHF altered responsiveness to Ca2+ (reduced Ca2+ affinity to troponin C) • Altered gene expression abnormal protein synthesis fatal myosin isoforms
Factors stimulate cell growth Cell deformation Stretch activated ion channels Cytoskeletal rearrangements (microtubules, desmin) Extracellular growth factors FGF (fibroblast growth factor) TGF (type transforming factor) Extracellular neurohormonal pharmacological modulators -adrenoceptor stimulants -adrenoceptor stimulants angiotensin II and endothelin thyroxin, insulin growth hormone (GS/TGF-I ratio) glucocorticoids cytokines TNF Intracellular energy deficit decreased high energy phosphates (ATP, CP) increased products of excessive energy utilisation (ADP, AMP, ceratine) Intracellular second messengers cAMP, cAMP-dependent kinases calcium and IP3/DG/PKC pathway Cellular protooncogens c-fos, c-myc, c-jun Cellular signalling factors citochrome-c and apoptotic protein activting factor-1 caspases protoapoptotic proteins (bax) Death receptors Fas TNF receptors
Genetic factors in the development of heart failure 1. Non-familial hypertrophic cardiomyopathy (NF-HCM) LV mass is partially determined by familial influence and 60 % of the variability can be explained by heritable factors. Local RAS gene polimorphism predisposition to hypertrophy. Genetic polimorphism in intron 16 of ACE gene, characterised by an insertion (I) or a deletion (D) of a 287-bp sequence. This ACE I/D polimorphism is strongly related to ACE plasma level and myocardial concentration. 2. Familial hypertrophic cardiomyopathy (FHC) Genetically heterogenous - 7 genes have been identified as responsible for the disease: 14q11-12 - b myosin heavy chain 1q3 - cardiac troponine T 15q2 - a-tropomyosin 11p11.2 - cardiac myosin binding protein c 12q - regulatory light chain of myosin 3p - essential light chain of myosin 19p13-q13 - cardiac troponine I
Catabolic/anabolic imbalance The general feature of neuroendocrine abnormalities: A catabolic/anabolic imbalance exists in HF. TNF is a key factor regulating energy metabolism, immune status, neuroendocrine and hormonal function. • Catabolic/anabolic status in CHF can be estimated by the cortisol/DHEA (dehydroepiandosterone) ratio. This ratio is highest in cachectic patients and correlates strongly with the degree of immune activation, represented by circulating TNF and soluble TNF receptor 1 and 2. • Cytokines induce programmed cell death (apoptosis) which is present in the skeletal musculature especially in cachectic patients. • Growth hormone (GH) - insulin like growth factor-I (IGF-I) axis is abnormal in severe CHF. In cachectic CHF patients GH is elevated and IGF-I is normal or low. • Insulin resistance is frequently observed in CHF. (Insulin is the strongest endogenous anabolic hormone which regulates the metabolic status of peripheral musculature. (Fasting insulin levels are only increased in non-cachectic patients. This might be due to a compensatory metabolic mechanism to overcome the insulin resistance). Use of insulin sensitizers might be useful! • Immune activation is present in CHF. TNF could be casual for the metabolic disturbances: • elevated metabolic rate • impaired tissue flow • altered fat and protein metabolism TNF is mainly elevated in cachectic CHF patients and it is the strongest predictor of the degree of weight loss.
Apoptosis in heart failure Apoptosis is an important mode of cell death (progressive loss of cardiac myocytes) in heart failure. AGII promotes apoptosis Apoptotic pathways: 1. Cytochrome c- is released in response to an apoptotic stimulus from the mitochondria. Cytochrome c, in the presence of dATP, forms an activation complex with apoptotic protein- activating factor-1 and caspase-9. This complex activates downstream caspases which leads to the final morphological and biochemical changes. This pathway is tightly regulated by a group of antiapoptotic proteins, such as Bcl-2 and proapoptotic proteins, such as Bax. Further regulation occurs downstream by various inhibitors of caspases. Bcl-2 is upregulated soon after coronary artery occlusion, espcially in the salvagable myocardium but is decreased in chronic HF induced by pressure overload. Apoptosis occurs in a high rate during reperfusion . The overexpression of BCL-2 effectively reduces reperfusion injury by reducing myocyte apoptosis. Bcl-2/Bax balance is important in the increased rate of apoptosis in cardiac myocytes.. 2. Death receptors (e.g. Fas, and TNF receptors) and caspase 8 also activate downstream caspases. Expression of Fas is upregulated in cardiac myocytes during ischaemia and heart failure. Antiapoptotic therapy includes: Beta adrenoceptor blockers: e.g. Carvedilol ACE inhibitors Caspase inhibitors Some hypertrophic signalling factors, such as cardiotrophin-1 via gp 130, insulin-like growth factor-1 via phosphoinositide-3-kinase, and calcineurin via the nuclear factor of activated T-cells seem to be protective.
Apoptotic stimulus citochrom c release from the mitochondrium activation complex caspase-cascade activation morphological and biochemical alterations Pprotein-activating factor caspase 9 Antiapoptotic protein Bcl-2 Proapoptotic protein Bax „death receptors” Fas és TNF
GOALS OF DRUG THERAPY FOR CHF The major goal of therapy is: to increase cardiac contractility (positive inotropic action) improve cardiac output to stop progression 1.) Improving the ability of the heart to meet the demands placed upon it (eg. by increasing contractility), or 2.) By reducing the demands being placed on the heart (eg. by reducing afterload with vasodilators) A: Drugs which enhance contractility of the failing myocardium a.) Cardiac glycosides b.) dopamine and dobutamine c.) PDE III inhibitors (amrinone, milrinone) B: Vasodilators To reduce preload and afterload a.) Venodilators (nitrites and nitrates) b.) Direct acting arterial dilators (hydralazine and minoxidil) c.) Alpha-adrenoreceptor blocking agents (prazosin) d.) Calcium antagonists (nifedipine) C: ACE inhibitors To reduce afterload and inhibit the progression of hypertrophy (gene expression ?) Captopril, Enalapril, Ramipril D: Antiarrhythmic agents To reduce irregular ventricular arrhythmias and prevent sudden death E: Diuretics Use to decrease edema, reduce blood volume. However, vigorous diuresis can be harmful (excessive reduction in preload which leads to a further decrease in CO).
Role of positive inotropic drugs in the treatment of CHF Positive inotropic agents are able to 1. Increase the extent and the speed of myocardial shortening (when preload, afterload, heart rate are kept constant). Act in normal myocardium, some play a physiologic role: NE, E. 2. Improve contractility of the failing heart during polonged administration Goals for use of positive inotropic drugs 1. Immediate lie-saving situations (after cardiac surgery, intensive care) i.v. DOPAMINE, DOBUTAMINE, DOPEXAMINE, ENOXIMONE, LEVOSIMENDAN, are useful if depression of myocardial function is thought to be reversible and is primarily related to abnormal excitation-contraction coupling 2. Chronic heart failre from NYHA II to NYHA IV they remain the part of the therapy despite full therapy with diuretics, vasodilators and ACE inhibitors. Aim to improve the symptoms and quality of life; if possible to improve survival. CARDIAC GLYCOSIDES, PDE INHIBITORS.
CARDIAC GLYCOSIDES Egyptans 3000 years ago. In the 18th century William Withering described the clinical effects of an extract of the foxglove plant (Digitalis purpurea). Chemistry All of the used cardiac steroids, or cardenolides combine a steroid nucleus with an unsaturated lactone ring at the 17 position and a series of sugars linked to carbon 3 of the nucleus. The lactone ring and the steroid nucleus are essential for activity. The pharmacological active principle is the genin or aglicone. Three aspects of this general structure are required for optimal activity: a.) the hydroxyl at position of 14 b.) the unsaturated (5 or 6 numbered) lactone ring at position of 17 c.) the cis relationship between rings C and D (all other natural steroids are trans) The sugars are not necessary for activity but greatly affect water solubility, the speed of onset, potency and duration of action of the drug. Sources of these drugs: white and purple foxglove (D lanata and D purpurea), Mediterranean sea onion (squill), Strophantus gratus, Oleander, lilly of the valley etc. Certain toads skin glands: bufadielonides (6 membered lactone ring)
PHARMACOLOGICAL ACTIONS 1.) POSITIVE INOTROPIC ACTION (force of myocardial contractility) They increase the force and velocity of cardiac contractions (dP/dt). Mechanism of action Inhibition of K+-Na+ATP-ase (membrane bound enzyme, associated with the "sodium pump") The therapeutic direct action: increase the intensity of the "activate state" of the contractile apparatus by increasing free Ca2+ concentration in the vicinity of the contractile proteins during systole) The facilitation of excitation-contraction coupling may be as a result of: 1.) Inhibition of Na+-K+ATP-ase reduced Na+ transport out increased [Na+]i (1) reduced normal transport of Ca2+ out (via Na+/Ca2+ exchange) increased [Ca2+]i (1/a) 2.) Facilitation of Ca2+ entry, through the voltage-gated Ca-channels, during the plateau phase of action potential (2). 3.) Increased release of stored Ca2+ from the SR (3). NOTE: Toxic effects are well correlated to inhibition of ATP-ase and to ‘calcium overload’. Loss of intracellular K+ (increase in [Na+], and increase in [Ca++]i) favours the induction of arrhythmias.
Haemodynamic effects of cardiac glycosides a.) Effects in patients with heart failure • Cardiac glycosides increases CO. • All of the other observed changes are secondary to this one effect. b.) Relationship of ventricular function N to A = reduction in myocardial contractility A to B = compensation (ie. increase in preload to increase output of failing heart) B to C = digitalis action (ie. increased myocardial contractility) C to D = reduction in heart size (ie. decreased preload) secondary to improved performance (CO) during digitalis treatment D to E = reduction in filling pressure with no positive inotropic intervention (eg. diuretics) c.) Effect of digitalis in normal patients (1) Myocardial contractility increases (2) Vascular tone increases (3) CO does not change or may even decrease
2. Indirect (vagal) electrophysiolcical effects a.) BRADYCARDIA: both direct and vagal effects Vagus effect is due to: • stimulation of the vagal nucleus • greater sensitivity of the heart to Ach This can be abolished by atropine or by vagotomy • In lower doses, cardioselective parasympathomimetic effects predominate • Cholinergic innervation: in the atria and AV node • Less indirect effect on Purkinje or ventricular function. In hear failure tachycardia can abolish automatically when CO is increased b.) SHORTENING OF THE REFRACTORY PERIOD (RP) OF ATRIAL MUSCLE • Speeding of atrial rate atrial flutter transfers to fibrillation c.) SLOWING CONDUCTION THROUGH THE AV NODE • Prolonged P-R interval (1o heart block) • Dropped beats (2o heart block) • Complete AV dissociation (3o heart block) • Slowing of ventricular rate during atrial flutter or fibrillation Since ventricular rate depens primarily on the activity of the AV node, prolongation of the RP of the AV node protects the ventricle from the rapid atrial impulses ventricular rate will be slowed
3. Direct electrophysiological effects (1) Atrial muscle • Early, brief prolongation of AP (increased membrane resistance), followed by • shortening of the AP (decreased membrane resistance; due to increased [Ca2+]i increased [K+]out). This results in AP shortening of atrial and ventricular refractoriness. (2) AV node • Slowed conduction, prolongation of RP (direct effect is synergistic with vagal effects). (3) Automaticity • Digitalis increases the automaticity in the latent pacemakers • It generates ”afterdepolarizations, afterpotentials" arrhythmias. • Slowing of intracardiac conduction (toxic doses) and increased automaticity leads to: - ES formation - AV junctional rhythm - bigeminy - VT, VF - asystole (cardiac standstill) Digitalis can cause virtually every variety of arrhythmia. Conduction disturbance (AV block): due to Na+ pump inhibition Arrhythmias: due to oscillatory afterdepolarizations (caused by overload of intracellular Ca2+).
Electrocardiographic effects ECG changes: ST-segment depression, inversion of T wave, PR prolongation, QT shortening. Induction or increase of U waves. These precede signs of toxicity such as bigeminal rhythm, ES, AV dissociation and ventricular arrhythmias. Ventricular arrhythmias (1) Cardiac glycosides are utilized as antiarrhythmic drugs: • supraventricular tachyarrhythmias (increase the RP of the AV node; flutter 2 :1; fibrillation 3 : 1) slower ventricular rate increase in CO (diastolic filling time increases) (2) Cardiac glycosides may cause virtually any type of arrhythmias (ventricular or supraventricular). • Inhibition of Na+-K+ATP-ase [Na]+i increases resting MP reduces • Increased rate of diatolic depolarization of the Purkinje cells • Decreased AV conduction (direct and indirect effects) • Abnormal automaticity (delayed afterdepolarizations) arrhythmias Vascular system • Direct constriction on arterial and venous smooth muscle increased TPR and BP (best seen after iv injection in normals) • Venoconstriction seen in CHF patients decreases after cardiac glycosides (cardiac function, compensatory sympathetic tone)
Gastrointestinal effects • GIT is the main extracardiac site of digitalis effect (unwanted side effects) anorexia, nausea, vomiting, diarrhea • These effects are partially due to the direct effects on the GIT or indirect; ie. stimulation of CNS, including chemoreceptor trigger zone CNS effects • Stimulates the vagal nucleus in the medulla slowing HR and increase in GIT motility. • Stimulates chemoreceptor emetic zone in the area postrema nausea and vomiting • Visual changes - changes in color vision • Neurological symptoms- headache, fatigue, disorientation, digitalis delirium seen particularly in elderly, rare convulsions, facial pain, similar to trigeminal neuralgia Other effects • Diuresis • due to increased cardiac function and circulation (renal blood flow) • inhibition of K+Na+ATP-ase in the kidneys • Interactions with K+, Ca2+ and Mg2+ • K+ and Ca2+ are antagonistic • K+ and digitalis (i) inhibit each-other's binding to Na+-K+ATP-ase, therefore, hyperkalemia reduces, hypokalemia faciltates the effects of cardiac glycosides •Ca2+ facilitates the toxic actions of digitalis • Mg2+ opposes the effect of Ca2+
Indications a.) Heart failure with atrial or supraventricular tachyarrhythmias (flutter or fibrillation) b.) Atrial flutter or fibrillation with rapid ventricular rate c.) Acute supraventricular tachycardia and decompensated heart d.) Prevention of atrial fibrillation and junctional tachycardia Contraindications a.) Hypertrophic cardiomyopathy (hypertrophic subaortic stenosis); cardiac glycosides increase the obrtruction against ejection, and inhibit relaxation b.) WPW syndrome (they enhance the redtrograde pulse conduction, provoke VT) c.) AV block Relative contraindications a.) If the decompensation is caused by: pericarditis, valvular stenosis, cor pulmonale b.) Hyperthyreosis (high CO syndrome) c.) Acute myocarditis d.) Acute myocardial infarction, ischaemia e.) Hypokalemia, renal insufficiency f.) Together with calcium antagonists, beta blockers, quinidine (reduce clearence of digitalis)
Toxic effects of digitalis Reason: calcium overload, N+K+ATP-ase inhibition. Toxicity is exacerbated by: • sympathomimetics • increase in calcium • decrease in magnesium • hypoxia • increased heart rate • potassium depletion Symptoms: extreme bradycardia, arrhythmias, anorexia, fatigue, headache, nausea, neuralgic pain and altered color vision (yellow hues) Treatment of toxicity • Discontinue cardiac glycosides • Correct precipitating factors (eg. electrolite disturbance) • Treat serious arrhythmias • K+ salts with normal renal function and constant monitoring • Antiarrhythmic drugs (Phenytoin, Lidocain) • Asystole may result in presence of complete heart block and abolition of ventricular arrhythmia • Steroid binding resins (primarily for digitoxin) and digoxin specific antibodies may be useful to aid drug removal
OTHER POSITIVE INOTROPIC DRUGS A: BETA ADRENERGIC RECEPTOR AGONISTS DOPAMINE, DOBUTAMINE, DOPEXAMINE • The positive inotropic action is accompanied only with little chronotropic activity or incerase in TPR • Use in acute heart failure (iv) due to AMI ISOPROTERENOL is NOT used in CHF (HR) ARAMINE (METARAMINOL), XAMOTEROL and METOPROLOL • Partial agonists, stimulate receptors positive inotropic action during long- term administration • receptor antagonists when sympathetic drive incerases (stress, exercise) • Improves LV diastolic function • They could be detrimental in NYHA IV • Indication: mild or moderate HF B: PDE INHIBITORS AMRINONE (INOCOR), MILRINONE (PRIMACOR), ENOXIMONE Mechanism of action • PDE inhibition cAMP Ca2+ influx through calcium channels • Increased release of Ca2+ from SR Result: positive inotropic action, balanced veno and arterial dilation Efficacy: • Negative during prolonged therapy in mild and severe HF • Proarrhythmic • No functional benefit, incerased mortality compared to digoxin Since in end-stage failure cAMP production is reduced (downregulation of receptors) PDE inhibitors and agonists are not adequately effective.
Preparations Narrow therapeutic range, small therapeutic index. LANOXICAPS (LANOXIN) - DIGOXIN LANATOZID C - duration is similar to digoxin, but poor oral absorpt OUABAIN (Strophantin) - short acting, only used experimentally ACETYLSTROPHANTIDIN - ultra short acting, only used experimentally ACIGOXIN (ACETYLDIGITOXIN) - lanatoside A glycoside; inj. tabl. CARDITOXIN (DIGITOXIN) - tabl. DIGOXIN - inj, solutio, tabl. ISOLANID (DESLANATOSID) - Lanatoside C glycoside, inj, tabl. TALUSIN (PROSCILLARIDIN) - tabl. DIGITALIS LEAF (whole leaf preparation) - duration is similar to digitalis but less potential Administration • Individual dosing ("titration”) to achieve adequate therapeutic effects and minimize undesirable side effects or toxicity. • Digitalizing dose and maintenance dose: Tradicional: large starting doses to achieve high plasma levels and tissue concentration, followed by smaller doses to maintain plasma levels. Modern: slower dosage is recommended (large starting doses only in emergency situation)
C: CALCIUM SENSITIZERS SULMAZOL, PIMOBENDAN, SIMENDAN, LEVOSIMENDAN Mechanism of action • Sensitisation of contractile proteins (troponin C) for calcium positive inotropic action • Inhibits PDEIII enzyme cAMP vasodilatation unwanted tachycardia arrhythmia generation D: VASODILATORS NITRATES (NITROGLYCERIN, NITROPRUSSID) CALCIUM ANTAGONISTS (NIFEDIPINE = CORINFAR = ADALAT) DIRECTLY ACTING VASODILATORS (HYDRELAYINE = DEPRESSAN, MINOXIDIL) ALFA-ADRENOCEPTOR BLOCKERS (PRASOZIN = MINIPRESS) Vasodilators therapy is advocated: • systemic resistance is increased in CHF • vasodilators (e.g. Nitrates) reduce LV filling pressure and increase CO However, most vasodilators are not selective the initial enthusiasm has waned • The immediate haemodynamic effects are not sustained in the long-term • They do not relate to long-term clinical improvement • They do not increase excersise capacity • There is no evidence that they alter mortality • Nitrates could be used to delay progression of myocardial damage
E: ACE INHIBITORS CAPTOPRIL (TENSIOMIN), ENALAPRIL (RENITEC) Plasma RAS maintains circulatory homeostasis during acute and subacute alterations in cardiac output Tissue RAS contributes to maintain homeostasis during impairment of CO 1. Activation of the RAS Occurs in response to reduction in cardiac output. Arterial constriction SABP and DABP Catecholamine release Angiotensin II. aldosteron secretion (adrenal cortex) • sodium retention • water retention • potassium excretion Increased blood pressure • water intake • vasopressin secretion • adrenocorticotropic hormon AGII binding to AT1 receptor leads to cardiac remodelling which includes: • upregulation of many early active cardiac myocyte genes • induction of late markers of cardiac hypertrophy (a-actin) and growth factors such as TGF • shift to fetal type myocardium AGII binding to AT2 receptor may oppose the effect of AGII on AT1 receptors
2. Hyperaldosteronemia AGII activates aldosterone secretion (+ decreases hepatic aldosterone clearance) Incerased aldosterone levels are indicators of HF (like the increased level of ANP) Elevated aldosterone leads to • myocardial fibrosis • sympathetic activation • BRS activation • magnesium loss • arrhythmias Angiotensin receptor blockade ACE inhibition fails to produce complete blockade of the RAS; ie. in response to the decrease in plasma AGII levels following ACE inhibition, the compensatory renin secretion rapidly restores AGII levels and this attenuates the effects of ACE inhibition. Similarly, cardiac ACE and chymase are specific AGII forming enzymes which are not abolished after chronic ACE inhibition. AT1 receptor antagonists: LOSARTAN, IBESARTAN, VALSARTAN, LISINOPRIL, CANDESARTAN Combined ACE inhibitor and AT1 receptor therapy: AT1 receptors are inhibited + the ACE inhibition leads to reduced sympathetic tone and generation of vasodilator kinins. In addition in this case, AGII acts on AT2 receptors which effect, together with the increase in kinin production, might be beneficial.
Efficacy: • reduction in symptoms of HF • increased exercise capacity • delay in progression of damage • reduction in mortality • act in all patients with mild to severe HF • reduction in preload (venodilatation) • reduction in afterload (arterial dilatation) • improvement in regional blood flow (renal vasodilatation) • improvement in coronary blood flow (due to reduced NE release) • reduction in sympathetic tone and arrhythmias • prevention of cardiac hypertrophy and dilatation • prevention of cardiac remodelling F: BETA RECEPTOR ANTAGONISTS Sympathetic stimulation is involved in: • LV remodelling • loss of myocardial cells • gene expression Increased sympathetic drive induces: 1. myocardial hypertrophy and fibroblast hyperplasia through stimulation of 1 and receptors 2. accelerated myocardial cell lost - apoptosis 3. NA may induce apoptosis through receptor activation (this can be blocked by CARVEDILOL) 4. changes in myocardial gene expression which results in progressive worsening in contractility (downregulation of the adult type, high ATP-ase activity -myosin heavy chain isoform and upregulation of the fetal type, low ATP-ase activity -myosin heavy chain isoform)
Beta-adrenoceptor blockers PROPRANOLOL, METOPROLOL, BUCINDOLOL, CARVEDILOL First generation: PROPRANOLOL (non selective) blocks all myocardial receptors and increases systemic vascular resistance Second generation: METOPROLOL and BISOPROLOL (1selective) produce lower reduction in cardiac index because they do not block cardiac 2 receptors and they have no effect on the 2-mediated vasodilatation. Metoprolol selectively upregulate 1 receptors and slightly improve maximal functional capacity Third generation: CARVEDILOL and BUCINDOLOL are non-selective or mildly selective agents. Their vasodilator (1 adrenoceptor blockade) activity may counteract their negative inotropic and 2 adrenoceptor blocking effects thus they do not worsen haemodynamics. They might yield a greater protection against the increased sympathetic drive. (i) They do not upregulate 1 receptors, a mechanism that may further reduce the sensitivity of the heart to sympathetic drive. (ii) They also block 2 receptors which because of 1 downregulation represents 40% of the total adrenergic receptors in patients with heart failure (dilated cardiomyopathy) and may mediate the cAMP-dependent effects of sympathetic stimulation even a greater extent than 1 receptors. (iii) Presynaptic 2 receptors facilitate NA release. Thus, only non-selective agents may decrease cardiac NA release. (iv) Cardiac 2 receptors may favour malignant tachyarrhythmias through cAMP.
CRITICAL EVALUATION OF DRUGS USED IN THE TREATMENT OF CHF Positive inotropic drugs 1. CARDIAC GLYCOSIDES (particularly digoxin) • Improvement in exercise tolerance (controlled trials: captopril-digoxin, milrinone- digoxin, xamoterol-digoxin) • In patients with sinus rhythm it’s efficacy is questionable • No improvement in NYHA IV. • Effect on cardiac mortality is controversial USEFUL BUT WEAK POSITIVE INOTROPIC DRUGS WITH LOW THERAPEUTIC INDEX 2. PDEIII INHIBITORS (AMRINONE, MILRINONE) • Efficacy during prolonged therapy in mild to severe HF is negative • Danger in arrhythmogenesis • Lack in functional benefit • Increased mortality (cp. to digoxin) • In end-stage failure (cAMP production is reduced) they are not effective 3. BETA-ADRENOCEPTOR PARTIAL AGONISTS (XAMOTEROL) • In normal subjects positive inotropic action • If sympahetic drive is increased (exercise or severe HF) acts as -blocker • Improves LV diastolic function • Improves symptoms and quality of life in mild and moderate HF 4. VASODILATORS • Most vasodilators are not selective • The immediate beneficial haemodynamic effects are not sustained in the long-term • Exercise capacity is not improved • There is no evidence that they alter mortality Nitrates could be used to delay progression of myocardial damage