Download

1 / 31
390 likes | 1.11k Views
Protein Threading. Zhanggroup 2003 10 22. Overview. Background protein structure protein folding and designability Protein threading Current limitations to protein threading. Computational complexity of certain formulations of the protein threading problem

E N D
Protein Threading Zhanggroup 2003 10 22
Overview • Background protein structure protein folding and designability • Protein threading • Current limitations to protein threading
Computational complexity of certain formulations of the protein threading problem • Performance of protein threading systems • References
Protein Structure • Primary, secondary, tertiary structure
Can only refer to the structure of a protein if a particular environment is assumed • solvent environment (aqueous trans-membrane ……) • temperature • pH etc • Different environments yield different structures or no stable structure at all
Proteins molecules are not completely rigid structures • kinetic energy energetic collisions with solvent molecules • vibrations sidechain conformational changes • flexible sections of the peptide chain • The native tertiary structure of a protein is thus an average
Protein Folding • Protein folding = searching for a conformation having minimum energy
Factors in protein folding • hydrophobic effects • electrostatic charges in residues • hydrogen bonding • Chaperonins,ribosomes
3 stages of folding • denatured unfolded state • molten globule state • native compact state • most proteins will return to their native state after forced denaturation
The Protein Folding Problem • Given a proteins amino acid sequence what is its tertiary structure • The protein folding problem is hard
Direct approach :molecular dynamics simulation • Simulate on an atomic level the folding of a single protein molecule • protein = thousands of atoms • solvent environment = hundreds to thousands of molecules => thousands of atoms
Sub-picosecond time scales • run the simulation for 1-5 seconds • We need another years of Moores law to make this computation feasible
Designability • A protein with a stable native state can not have another low-energy state nearby in conformational space • A structure is highly designable if its minimum energy state has no low-energy neighbours
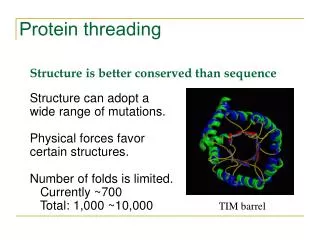
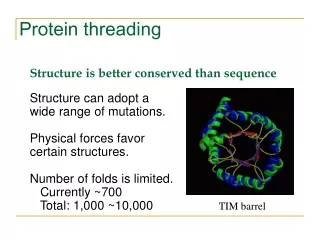
Protein Threading • inverse protein folding problem: given • a tertiary structure, find an amino acid sequence that folds to that structure • Protein threading: given a library of possible protein folds and an amino acid sequence find the fold with the • best sequence -> structure alignment (threading)
Evolution depends on designability to preserve function under mutation • Estimate only different protein structures exist in nature (Chothia,1992)
four components • a library of protein folds (templates) • a scoring function to measure the fitness of a sequence -> structure alignment • a search technique for finding the best alignment between a fixed sequence and structure
a means of choosing the best fold from among the best scoring alignments of a sequence to all possible folds
Scoring Schemes for Sequence->Structure Alignments • The scoring scheme for a particular threading of a sequence onto a structure measures the degree to which
environmental preferences are satisfied • Different amino acid types prefer different environments e.g. • structural preferences: • in helix • in sheet • not exposed to solvent • pairwise interactions with neighbouring amino acids
Formal Statement of the ProteinThreading Problem • C is a protein core having m segments Ci representing a set of contiguous amino acids Let ci be the length of Ci • Sequence a = a1a2…an of amino acids
Current limitations to protein threading • Statistical problems • Definition of neighbor and /or pairwise contact environments: • energetic neighbor ? contact neighbor
Computational Complexity of Finding an Optimal Alignment • The complexity of the protein threading problem depends on whether: • Variable-length gaps are allowed in alignments • the scoring function for an alignment incorporates pairwise interactions between amino acids
Property(I) makes the search space exponential in size to the length of the sequence • Property(Ii) forces a solution to take non-local effects into account
Any protein threading scheme with both properties is NP-complete (3-SAT Lathrop 1994) (MAX-CUT Akutsu,Miyano 1999)
Thus all protein threading approaches can be divided into four groups: 1 no variable length gaps allowed 2 no pairwise interactions considered in scoring function 3 no optimal solution guarantee 4 exponential runtime
Performance of Protein Threading Systems CASP1(1994) CASP2(1996) CASP3(1998): Critical Assessment of Structure Prediction meetings protein threading methods have consistently been the winners success depends on structural similarity of target to known structures successful even when target sequence and library sequence have low homology
Much room for improvement in all areas of protein threading e.g.: algorithms for searching the threading space reliable biologically accurate scoring functions