Download

1 / 66
690 likes | 750 Views
RNA sequencing, transcriptome and expression quantification. Henrik Lantz, BILS/SciLifeLab. Lecture synopsis. What is RNA-seq? Basic concepts Mapping-based transcriptomics (genome -based) De novo based transcriptomics (genome-free) Expression counts and differential expression

E N D
RNA sequencing, transcriptome and expression quantification Henrik Lantz, BILS/SciLifeLab
Lecture synopsis • What is RNA-seq? • Basic concepts • Mapping-based transcriptomics (genome -based) • De novo based transcriptomics (genome-free) • Expression counts and differential expression • Transcript annotation
RNA-seq DNA Exon Intron Exon Intron Exon Intron Exon UTR UTR AG GT AG GT GT AG ATG Start codon TAG, TAA, TGA Stop codon Transcription Pre-mRNA UTR UTR AA AAAAA ATG Start codon TAG, TAA, TGA Stop codon Splicing mRNA UTR UTR AAAAAAAAA ATG Start codon TAG, TAA, TGA Stop codon Translation
Overview of RNA-Seq Reconstruct original full-length transcripts From: http://www2.fml.tuebingen.mpg.de/raetsch/members/research/transcriptomics.html
Common Data Formats for RNA-Seq FASTA format: >61DFRAAXX100204:1:100:10494:3070/1 AAACAACAGGGCACATTGTCACTCTTGTATTTGAAAAACACTTTCCGGCCAT FASTQ format: @61DFRAAXX100204:1:100:10494:3070/1 AAACAACAGGGCACATTGTCACTCTTGTATTTGAAAAACACTTTCCGGCCAT + ACCCCCCCCCCCCCCCCCCCCCCCCCCCCCBC?CCCCCCCCC@@CACCCCCA Quality values in increasing order: !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~ You might get the data in a .sff or .bam format. Fastq-reads are easy to extract from both of these binary (compressed) formats!
Insert size Insert size Read 1 DNA-fragment Read 2 Adapter+primer Inner mate distance
Paired-end gives you two files FASTQ format: @61DFRAAXX100204:1:100:10494:3070/1 AAACAACAGGGCACATTGTCACTCTTGTATTTGAAAAACACTTTCCGGCCAT + ACCCCCCCCCCCCCCCCCCCCCCCCCCCCCBC?CCCCCCCCC@@CACCCCCA @61DFRAAXX100204:1:100:10494:3070/2 ATCCAAGTTAAAACAGAGGCCTGTGACAGACTCTTGGCCCATCGTGTTGATA + _^_a^cccegcgghhgZc`ghhc^egggd^_[d]defcdfd^Z^OXWaQ^ad
Transcript Reconstruction from RNA-Seq Reads Nature Biotech, 2010
Transcript Reconstruction from RNA-Seq Reads MAPPING TopHat
Transcript Reconstruction from RNA-Seq Reads MAPPING TopHat Cufflinks
Transcript Reconstruction from RNA-Seq Reads MAPPING Trinity The Tuxedo Suite: End-to-end Genome-basedRNA-Seq Analysis Software Package TopHat GMAP Cufflinks
Transcript Reconstruction from RNA-Seq Reads MAPPING Trinity TopHat Cufflinks
Transcript Reconstruction from RNA-Seq Reads MAPPING Trinity TopHat GMAP Cufflinks
Transcript Reconstruction from RNA-Seq Reads End-to-end Transcriptome-basedRNA-Seq Analysis Software Package Trinity GMAP
Basic concepts of mapping-based RNA-seq - Coverage Reads 5x Coverage 2x Mapping Overlapping reads Reference genome Coverage = number of reads at a certain position Higher coverage in RNA-seq=>higher chance of sequencing low-abundance transcripts
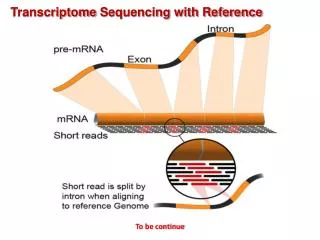
Basic concepts of mapping-based RNA-seq - Spliced reads DNA Exon Intron Exon Intron Exon Intron Exon UTR UTR AG GT AG GT GT AG ATG Start codon TAG, TAA, TGA Stop codon Transcription Pre-mRNA UTR UTR AA AAAAA ATG Start codon TAG, TAA, TGA Stop codon Splicing mRNA UTR UTR AAAAAAAAA ATG Start codon TAG, TAA, TGA Stop codon Translation
Pre-mRNA DNA Exon Intron Exon Intron Exon Intron Exon UTR UTR GT GT GT ATG Start codon TAG, TAA, TGA Stop codon Transcription Pre-mRNA UTR UTR ATG Start codon TAG, TAA, TGA Stop codon Splicing mRNA UTR UTR ATG Start codon TAG, TAA, TGA Stop codon Translation
Overview of the Tuxedo Software Suite Bowtie (fast short-read alignment) TopHat (spliced short-read alignment) Cufflinks (transcript reconstruction from alignments) Cuffdiff (differential expression analysis) CummeRbund (visualization & analysis)
Alignments are reported in a compact representation: SAM format 0 61G9EAAXX100520:5:100:10095:16477 1 83 2 chr1 3 51986 4 38 5 46M 6 = 7 51789 8 -264 9 CCCAAACAAGCCGAACTAGCTGATTTGGCTCGTAAAGACCCGGAAA 10 ###CB?=ADDBCBCDEEFFDEFFFDEFFGDBEFGEDGCFGFGGGGG 11 MD:Z:67 12 NH:i:1 13 HI:i:1 14 NM:i:0 15 SM:i:38 16 XQ:i:40 17 X2:i:0 SAM format specification: http://samtools.sourceforge.net/SAM1.pdf
Alignments are reported in a compact representation: SAM format (read name) 0 61G9EAAXX100520:5:100:10095:16477 1 83 2 chr1 3 51986 4 38 5 46M 6 = 7 51789 8 -264 9 CCCAAACAAGCCGAACTAGCTGATTTGGCTCGTAAAGACCCGGAAA 10 ###CB?=ADDBCBCDEEFFDEFFFDEFFGDBEFGEDGCFGFGGGGG 11 MD:Z:67 12 NH:i:1 13 HI:i:1 14 NM:i:0 15 SM:i:38 16 XQ:i:40 17 X2:i:0 (FLAGS stored as bit fields; 83 = 00001010011 ) (alignment target) (position alignment starts) (Compact description of the alignment in CIGAR format) (read sequence, oriented according to the forward alignment) (base quality values) (Metadata) SAM format specification: http://samtools.sourceforge.net/SAM1.pdf
Alignments are reported in a compact representation: SAM format (read name) 0 61G9EAAXX100520:5:100:10095:16477 1 83 2 chr1 3 51986 4 38 5 46M 6 = 7 51789 8 -264 9 CCCAAACAAGCCGAACTAGCTGATTTGGCTCGTAAAGACCCGGAAA 10 ###CB?=ADDBCBCDEEFFDEFFFDEFFGDBEFGEDGCFGFGGGGG 11 MD:Z:67 12 NH:i:1 13 HI:i:1 14 NM:i:0 15 SM:i:38 16 XQ:i:40 17 X2:i:0 (FLAGS stored as bit fields; 83 = 00001010011 ) (alignment target) (position alignment starts) Still not compact enough… Millions to billions of reads takes up a lot of space!! Convert SAM to binary – BAM format. (Compact description of the alignment in CIGAR format) (read sequence, oriented according to the forward alignment) (base quality values) (Metadata) SAM format specification: http://samtools.sourceforge.net/SAM1.pdf
Samtools • Tools for • converting SAM <-> BAM • Viewing BAM files (eg. samtools view file.bam | less ) • Sorting BAM files, and lots more:
Text-based Alignment Viewer % samtoolstviewalignments.bamtarget.fasta
Transcript Reconstruction Using Cufflinks From Martin & Wang. Nature Reviews in Genetics. 2011
Transcript Reconstruction Using Cufflinks From Martin & Wang. Nature Reviews in Genetics. 2011
Transcript Reconstruction Using Cufflinks From Martin & Wang. Nature Reviews in Genetics. 2011
Transcript Reconstruction from RNA-Seq Reads Trinity The Tuxedo Suite: End-to-end Genome-basedRNA-Seq Analysis Software Package TopHat GMAP Cufflinks
Transcript Reconstruction from RNA-Seq Reads End-to-end Transcriptome-basedRNA-Seq Analysis Software Package Trinity GMAP
De novo transcriptome assembly No genome required Empower studies of non-model organisms • expressed gene content • transcript abundance • differential expression
The General Approach to De novo RNA-Seq AssemblyUsing De Bruijn Graphs
Sequence Assembly via De Bruijn Graphs From Martin & Wang, Nat. Rev. Genet. 2011
Contrasting Genome and Transcriptome Assembly Transcriptome Assembly Genome Assembly • Uniform coverage • Single contig per locus • Double-stranded • Exponentially distributed coverage levels • Multiple contigs per locus (alt splicing) • Strand-specific
Trinity Aggregates Isolated Transcript Graphs Genome Assembly Single Massive Graph Trinity Transcriptome Assembly Many Thousands of Small Graphs Ideally, one graph per expressed gene. Entire chromosomes represented.
Trinity – How it works: Transcripts + Isoforms de-Bruijn graphs RNA-Seqreads Linear contigs Thousands of disjoint graphs