Download

1 / 51
570 likes | 722 Views
Simulations of Peptide Aggregation. Joan-Emma Shea Department of Chemistry and Biochemistry The University of California, Santa Barbara. Protein and Peptide Aggregation. PROTEIN AGGREGATION AND DISEASE. Parkinson. Alzheimer. Prion (“Mad Cow”). Huntington.

E N D
Simulations of Peptide Aggregation Joan-Emma Shea Department of Chemistry and Biochemistry The University of California, Santa Barbara
PROTEIN AGGREGATION AND DISEASE Parkinson Alzheimer Prion (“Mad Cow”) Huntington Proteins not associated with a specific disease can also aggregate to form amyloid fibrils
Nano-tube and nano-sphere fabrication using aromatic di-peptides. [Gazit et al., Nano Lett, 4 (2004) 581] APPLICATION TO BIOMATERIALS Use of peptides to form nanoscale-ordered monolayers, COSB 2004, 14: 480
Time scales Side-chain rotations Loop closure Protein aggregation Protein folding Folding of -hairpins Helix formation
MULTISCALE APPROACH All-atom: Molecular Dynamics (MD) with IMPLICITsolvent Off-lattice minimalist: Langevin dynamics COARSE GRAINING All-atom: Molecular Dynamics (MD) With EXPLICIT Solvent
1. Dimerization Mechanisms of 4 tetrapeptides KXXE Fibrils of KFFE 2. Design of Inhibitors of aggregation of the Alzheimer Amyloid-beta (A) peptide.
Fibrils! KXXE peptides Tjernberg et al. , JBC 277 (2002) 43243 KAAE KLLE KVVE KFFE
PEPTIDE MODEL CHARMM19 Force Field and Generalized Born Implicit Solvent R ep=1 kCal/mol, s=1Å, R=17Å Confining Sphere:
T1 NVT NVT T2 TN Simulation protocol: Replica Exchange MD6 replicas for monomer: total simulation time: 40 ns12 replica for dimers: total simulation time: 400 ns
Heterogeneous dimers E (kCal/mol) RMSD (Å)
Thermodynamic stability of dimers: KFFE> KVVE > KLLE > KAAE Association temperature Ta
Propensity for structure Salt bridges Hydrophobic contacts Aromatic interactions What determines transition temperature Ta?
T=325K T=240K T=275K Free energy as a function of interaction energy and radius of gyration T=285K Rg (Å) U (kCal/mol) U U U
T=325K T=240K T=275K Free energy as a function of interaction energy and radius of gyration T=285K Most favorable Rg (Å) Interaction energy: KFFE > KLLE ~ KVVE > KAAE DU (kCal/mol) DU Least favorable U U
Entropy loss due to dimerization Monomers, Rg>5A -strand Random Coil Helix
Entropy loss due to dimerization Monomers, Rg>5A -strand Random Coil Helix
Entropy loss due to dimerization Monomers, Rg>5A Dimers, Rg<5A -strand Random Coil Helix
Entropy loss due to dimerization Monomers, Rg>5A Dimers, Rg<5A -strand Random Coil -strand basin more populated for monomer of KVVE than KLLE: SL > SV Helix
Energetic effect Entropic effect Association temperature
FREE ENERGY BARRIER FOR KLLE Kinetic accessibility of dimers KFFE KLLE KLLE KFFE Rg (Å) E (Kcal/mol) E (Kcal/mol) “DOWNHILL” DIMERIZATION FOR KFFE
Different mechanisms of dimerization KFFE KLLE Rg over C Rg over PHE atoms Rg over LEU atoms PHE-PHE come together first (stabilized by vdw interactions), followed by the overall collapse of the structure with formation of peptide backbones contacts . LEU side chain formation and overall collapse with formation of peptide backbone interactions occur simultaneously.
CONCLUSIONS 1. for dimerization of the KXXE peptides 2. Strong sequence dependence of free energy landscapes for dimerization, with KFFE experiencing a barrierless transition. 3. KFFE dimers are the most thermodynamically stable and kinetically accessible. 4. Dimer trends match experimental trends observed for fibrils.
LARGER SYSTEMS: OLIGOMERIZATION OF THE ALZHEIMER AMYLOID BETA (A) PEPTIDES Aβ40: DAEFRHDSGYEVHHQ16KLVFFA22EDVGSNKGAIIGLMVGGVV Aβ42: DAEFRHDSGYEVHHQ16KLVFFA22EDVGSNKGAIIGLMVGGVVIA AMYLOID BETA (A) PEPTIDES AGGREGATE TO FORM TOXIC OLIGOMERS AND FIBRILS
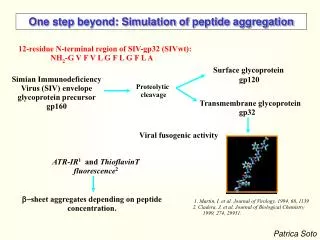
FRAGMENT 25-35 OF THE (A) PEPTIDE APPEARS TO BE TOXIC IN MONOMERIC, SMALL OLIGOMERIC AND FIBRILLAR FORMS Aβ40: DAEFRHDSGYEVHHQKLVFFAEDV25GSNKGAIIGL35MVGGVV NO STRUCTURE OF THE MONOMERIC PEPTIDE AVAILABLE IN AQUEOUS SOLVENT PEPTIDE ADOPTS A HELICAL STRUCTURE IN APOLAR ORGANIC SOLVENT (SUCH AS HEXAFLUOROISOPROPANOL HFIP)
EFFECTS OF SOLVENT ON FREE ENERGY LANDSCAPE OF THE MONOMER REPLICA EXCHANGE SIMULATION IN: HFIP/WATER CO-SOLVENT PURE WATER GROMOS96 FORCE FIELD, EXPLICIT SOLVENT 40 REPLICAS, 16 NS EACH, TOTAL SIMULATION TIME OF 640 NS
N C N C N C N C FREE ENERGY SURFACE IN HFIP/WATER COSOLVENT: HELIX STABILIZATION T=300 K 45 % 8%
HFIP DISPLACES WATER NEAR THE PEPTIDE, AND FORMS A “COAT” AROUND THE PEPTIDE
C N N N C C C N N C FREE ENERGY SURFACE IN PURE WATER FORMATION OF COLLAPSED-COILS and b-HAIRPINS T=300K
TWO TYPES OF ORDERED DIMERS Experimentally: two different protofilament of diameters: 1.41 +/- 0.48 nm 3.58 +/- 1.53 nm
PEPTIDE INHIBITORS OF ALZHEIMER A AGGREGATION Alzheimer’s disease (AD) is a neurodegenerative disease of the central nervous system.
ALZHEIMER DISEASE IS CHARACTERIZED BY THE PRESENCE OF NEUROFIBRILLAR TANGLES AND AMYLOID PLAQUES IN THE BRAIN
AMYLOID PLAQUES CONSIST OF AMYLOID BETA (A) PEPTIDES GENERATED FROM THE PROTEOLYTIC CLEAVAGE OF THE APP TRANSMEMBRANE PROTEIN Aβ40: DAEFRHDSGYEVHHQ16KLVFFA22EDVGSNKGAIIGLMVGGVV Aβ42: DAEFRHDSGYEVHHQ16KLVFFA22EDVGSNKGAIIGLMVGGVVIA
Aβ40: DAEFRHDSGYEVHHQ16KLVFFA22EDVGSNKGAIIGLMVGGVV Aβ42: DAEFRHDSGYEVHHQ16KLVFFA22EDVGSNKGAIIGLMVGGVVIA In healthy individuals, the A peptides are broken down and eliminated. In AD, these peptides self-assemble into amyloid fibrils Fibril Amyloid Plaques
Both small soluble oligomers and fibrils appear to be toxic to cells. Misfolded Protein Fibril Native Protein Soluble Oligomer Protofibrils
After Incubation with A(16-20)m peptides Fibrils of A(1-40) peptides Meredithand co-workers, J. Pep. Res. (2002) 60, 37-55 N-METHYLATED PEPTIDE INHIBITORS • N-methylated A(16-20)m peptides can: • prevent the aggregation of full length A peptide • disassemble existing fibrils and possibly small oligomers. 16K(me)LV(me)FF Aβ40: DAEFRHDSGYEVHHQ16KLVFFA22EDVGSNKGAIIGLMVGGVV
MODEL SYSTEM Antiparallel arrangements from solid state NMR Tycko et al. Biochemistry, 39 (45), 13748 -13759, 2000 Fragment A(16-22) KLVFFAE aggregates to form fibrils Aβ40: DAEFRHDSGYEVHHQ16KLVFFA22EDVGSNKGAIIGLMVGGVV
A(16-22) KLVFFAE PROTOFIBRIL INITIAL STRUCTURE: Two parallel bilayers Peptides in layer antiparallel lys16 and glu22 point to solvent leu17, phe19, ala21 point inside core
A(16-22) KLVFFAE PROTOFIBRIL INITIAL STRUCTURE: Two parallel bilayers Peptides in layer antiparallel lys16 and glu22 point to solvent leu17, phe19, ala21 point inside core GROMOS96 FORCE FIELD EXPLICIT SPC WATER (23000 atoms) REACTION FIELD/ PME TWO 20 NS SIMULATIONS
REPRESENTATIVE A(16-22) PROTOFIBRIL (LAST 7 NS OF SIMULATIONS) Distance between bilayers: 0.93 nm (Tycko: 0.99nm) Distance between peptides: 0.44-0.52 nm (Tycko: 0.47 nm)
Structure of N-methylated A(16-20)m Inhibitor Peptide Replica Exchange Molecular Dynamics, 30 replicas, 20 ns per replica A(16-20)m more rigid than A(16-22), with -strand content: This pre-organization may allow A(16-20)m to successfully compete with free A(16-22) for binding to fibril.
A(16-20)m with N-methyl groups pointing toward fibril A(16-20)m with N-methyl groups pointing away fibril Interaction of A(16-20)m Inhibitor Peptide with protofibril INITIAL STRUCTURE
A(16-20)m with N-methyl groups pointing toward fibril A(16-20)m with N-methyl groups pointing away fibril Forms hydrogen bonds with fibril (antiparallel) Intercalates between layers Interaction of A(16-20)m Inhibitor Peptide with protofibril AFTER 50 ns
POSSIBLE MECHANISM OF FIBRIL DISRUPTION Inhibitor drifts from edge of fibril to side and inserts in fibril (between strands 6 and 7) with Lys pointing to solvent and hydrophobic residues inserted in fibril.
POSSIBLE MECHANISM OF INHIBITION OF FIBRIL GROWTH Inhibitor forms hydrogen bond with fibril, with antiparallel alignment, possibly preventing additional A(16-22) peptides from binding.
DESTABILIZATION OF FIBRIL WHEN INHIBITOR INSERTED IN FIBRIL Inhibitor inserted in fibril affects the “twist” of the fibril and the distance between strands.
CONCLUSIONS AND FUTURE DIRECTIONS • Results suggest possible mechanisms of fibril • inhibition and disassembly • Extend the simulations to consider other orientations • of the inhibitor peptides • Study and design new inhibitors
ACKNOWLEDGEMENTS Dr. A. Baumketner Dr. G. Wei Dr. P. Soto Collaborator: Prof. Stephen Meredith, University of Chicago Other group members: M. Friedel, M. Griffin, A. Jewett, W. B. Lee and E. Zhuang Funding: NSF Career, David and Lucile Packard Foundation, A. P. Sloan Foundation, Army Research Office.