Download

1 / 93
950 likes | 1.14k Views
A Comprehensive Bioinformatics Study Of The Interaction Between Peripheral Proteins And Membrane. PhD Dissertation Defense Nitin Bhardwaj Dept of Bioengineering (UIC) Sept 5 th 2007. Talk Outline. Introduction/Motivation Problem statement Structure-based prediction

E N D
A Comprehensive Bioinformatics Study Of The Interaction Between Peripheral Proteins And Membrane PhD Dissertation Defense Nitin Bhardwaj Dept of Bioengineering (UIC) Sept 5th 2007
Talk Outline • Introduction/Motivation • Problem statement • Structure-based prediction • Black-box nature of classifiers • Integration of various techniques • Unavailability of an exclusive resource • Methodology development • Supervised machine learning • Unsupervised machine learning • MD simulation/Docking/Electrostatics with experimental techniques • Results • Conclusion/Outlook
Talk Outline • Introduction/Motivation • Problem statement • Structure-based prediction • Sequence-based prediction • Black-box nature of classifiers • Integration of various techniques • Unavailability of a dedicated resource • Methodology development • Supervised Machine learning • Unsupervised machine learning • MD simulation/Docking/Electrostatics with experimental techniques • Results • Conclusion/Outlook
Introduction • Cells continuously receive information ('signals') from their environment/neighbors • Respond and coordinate cellular changes. Control of fundamental processes: cell proliferation, metabolism, survival • Extracellular signals through ligand-receptor binding followed by activation of signaling pathways involving phosphorylation/dephosphorylation, various kinds of interactions (Picture from Cell Biology Project, Dept of Biochem at Univ of Arizona)
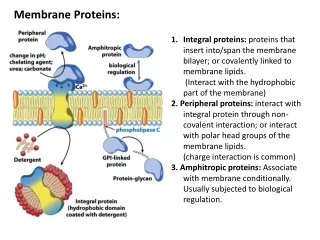
Introduction • Process of cellular signaling involves complex arrays of protein-protein and protein-lipid interactions • A large number of cytosolic proteins recruited to different cellular membranes, called as peripheral proteins • Not restricted to canonical signaling mechanisms; play roles in membrane trafficking and anchoring cytoskeletal structures
Introduction Intracellular Extracellular From the webpage for BIOS 100 at UIC (Summer 2006)
Introduction • Peripheral proteins bind the membrane mostly in a reversible manner using different strategies. • Contain one or more modular domains specialized in lipid binding called membrane-targeting domains • Include C1, C2, PH, FYVE, PX, ENTH, ANTH, BAR, FERM, and tubby domains.
Mechanisms of binding cPLA2-C2 domain epsin-ENTH domain β-subunits of G-proteins PKCγ-C1 domain PH domains
Introduction/Motivation • Many diseases caused by aberrant molecular changes affecting signal transduction, and can now be treated by drugs that target signaling proteins such as kinases • Over 400 inflammatory, cardiovascular and neuropsychiatric human diseases have been linked to defects in signal transduction pathways • Cancer/AIDS associated with signal-transduction proteins
Introduction/Motivation • In cancers, overproduction of a phospholipid, phosphatidylinositol (3,4,5) trisphosphate (PIP3), by phosphatidylinositol 3-kinase (PI3K) a common signal • PI3K pathway regulates various cellular processes, such as proliferation, apoptosis and through downstream action of AKT and mediates tumorogenesis • Activation of AKT initiated by membrane translocation by an interaction between its PH domain and PIP3. Regulation of AKT activity (Vivanco, I., Sawyers, C., Nature Reviews Cancer, 2002, 2 , 489)
Introduction/Motivation • During late phase of HIV type 1 (HIV-1) replication, newly synthesized retroviral Gag proteins targeted to the plasma membrane • Colocalize at lipid rafts and assemble into immature virions. • Ability of HIV-1 Gag to colocalize at specific subcellular membranes essential for viral replication Predicted Membrane-binding model of Gag (Saad, J. et al 2006 PNAS, 103 (30), 11364)
Talk Outline • Introduction/Motivation • Problem statement • Structure-based prediction • Sequence-based prediction • Black-box nature of classifiers • Integration of various techniques • Unavailability of a dedicated resource • Methodology development • Supervised Machine learning • Unsupervised machine learning • MD simulation/Docking/Electrostatics with experimental techniques • Results • Conclusion/Outlook
Structure-based Prediction • Do not have canonical trans-membrane segments • Experiments prohibitively time-consuming and expensive • Database searching does not always render credible results, eg, FYVE • Proteins with similar folds show different properties, eg. PH • Fast/efficient computational method not solely based on sequence homology or tertiary structural similarity • No previous attempt towards computational identification
Sequence-based Prediction • 3D structures of all proteins in the proteome of commonly studied organisms not available • For genome-wide prediction of peripheral proteins, need sequence-based prediction protocols that can give reasonable precursor performance • Broaden the prediction of peripheral proteins to that based on the features derived from their sequences
Sequence-based Prediction • No large and well-defined set of negative cases • No experiments to identify proteins that “do not bind” membranes • Conventional ML algorithms need both positive and negative sets
Black-box nature of classifiers • Nature of the classification is still black-box type; the underlying distribution of the features is unknown • Set of simple rules that could be interpreted in the biological context of these proteins: Classification trees • Goals: • Knowledge Mining ? Classifier • To identify binding and non-binding cases from the same domain
Integration of various techniques • An atomic-level analysis of mechanistic rules and conditions governing the association of these proteins with the membranes using molecular dynamics (MD) simulation • Docking • Electrostatics • Use these techniques to illuminate key details into the protein-membrane interaction such as favorable conditions, binding lipid partners, important cofactors, binding orientation, etc.
Unavailability of an exclusive Resource • Several resources available for protein-protein interactions (BIND, DIP, MIPS), Protein-DNA interaction (DPInteract, ProNIT), GPCRs, p53, protein-ligand interactions. • No specialized/comprehensive peripheral proteins resource • With ever increasing data available for the lipid-binding and membrane targeting domains, high quality dedicated and organized resource for these domains. • Created the only publicly-available resource for membrane-targeting domains
Talk Outline • Introduction/Motivation • Problem statement • Structure-based prediction • Sequence-based prediction • Black-box nature of classifiers • Integration of various techniques • Unavailability of a dedicated resource • Methodology development • Supervised Machine learning • Unsupervised machine learning • MD simulation/Docking/Electrostatics with experimental techniques • Results • Conclusion/Outlook
Supervised-machine learning • Problem: given training data, produce a classifier which maps an object to its classification label . The xivalues are typically vectors of the form <xi,1, xi,2, ..., xi,n> whose components are discrete or real values. • Given a set of training examples, a learning algorithm outputs a classifier. Given new x values, predicts the corresponding y values. • For example, while filtering spam, x: a representation of an email (the subject, body, etc) and y: {"Spam“, "Non-Spam”}.
Structure-based Prediction Overall strategy (N. Bhardwaj, R.V. Stahelin, R.E. Langlois, W. Cho, and H. Lu. 2006.J Mol Biol359: 486-495)
Unsupervised-Machine learning • Where examples from one of the classes or class labels are missing • Semi-supervised or partially-supervised • For peripheral domains, a well defined set of domains which are known to bind the membranes + a much larger set of proteins whose affinities have not been measured (unlabeled) • Supervised machine learning • Implemented Positive-Unlabeled(PU) learning which starts with two sets: a well-defined positive set, and a much larger set with unlabeled examples.
Knowledge Mining • To remove the black-box nature of the classifiers, “classification tree” • Put forth some biological rules that will be evaluated in the biological context of these proteins. • A rule is defined as a path from the root to a leaf. charge < 4.5 Red: Non-binding Black: Binding N Y -0.7 +0.2 Tyr > 3 Trp < 3 N N Y Y +0.4 -0.2 +0.3 -0.3 1abc 1def1ghi 1jkl 21 32 4 7 11 5 3 45 (N. Bhardwaj, and H. Lu. in preparation)
Knowledge Mining • Two sets of graphical models depicting the rules are put forward: one set for all membrane-targeting domains combined together, and one set specific to each domain. • Much more difficult than the ones handled in previous works (N. Bhardwaj, and H. Lu. in preparation)
Talk Outline • Introduction/Motivation • Problem statement • Structure-based prediction • Sequence-based prediction • Black-box nature of classifiers • Integration of various techniques • Unavailability of a dedicated resource • Methodology development • Supervised Machine learning • Unsupervised machine learning • MD simulation/Docking/Electrostatics with experimental techniques • Results • Conclusion/Outlook
Results: Structure-based PredictionGoal: To identify peripheral proteins on the basis of their structure-derived features
Structure-based Prediction • Dataset • 40 peripheral proteins from all domains known • 230 non-binding proteins • Descriptors • Net charge (anionic membrane) • Size of the cationic patches • ‘Overall’ and ‘surface’ amino acid composition (overall composition for initial recruitment, surface composition important for formation of more specific complex)
Structure-based Prediction • Distribution of descriptors (N. Bhardwaj, R.V. Stahelin, R.E. Langlois, W. Cho, and H. Lu. 2006.J Mol Biol359: 486-495)
Structure-based Prediction • Distribution of patches • In four cases, two of the cationic patches directly interact with the membrane; in one case only the largest patch in contact with the membrane ANTH domain PKCα C2 domain β-spectrin PH domain Cytosolic phospholipase A2 C2 domain ENTH domain (N. Bhardwaj, R.V. Stahelin, R.E. Langlois, W. Cho, and H. Lu. 2006.J Mol Biol359: 486-495)
Structure-based Prediction • Distribution of descriptors Surface Overall Binding proteins Non-binding proteins Astral-40 Database Ratio (N. Bhardwaj, R.V. Stahelin, R.E. Langlois, W. Cho, and H. Lu. 2006.J Mol Biol359: 486-495)
Structure-based Prediction • Performance of the protocol T=True, F=False, P=Positives, N=Negatives (N. Bhardwaj, R.V. Stahelin, R.E. Langlois, W. Cho, and H. Lu. 2006.J Mol Biol359: 486-495)
Prediction of Membrane Binding Properties of C2 Domains • Among four novel PKC C2 domains, membrane binding affinities of PKCδ-C2 and PKCε-C2 remain controversial, whereas those of PKCη-C2 and PKCθ-C2 have not been tested. • Computed all aforementioned features for four C2 domains • SVM classified PKCδ-C2, PKCε-C2, and PKCη-C2 as non-binding and PKCθ-C2 as a membrane-binding peripheral protein. 51.2% PKC-δ (1bdy) PKC-θ 50.1% PKC-ε (1gmi) PKC-η (N. Bhardwaj, R.V. Stahelin, R.E. Langlois, W. Cho, and H. Lu. 2006.J Mol Biol359: 486-495)
Validation of Membrane Binding Properties of C2 Domains • Dr Stahelin and Dr Cho measured the binding of the four C2 domains to phospholipid vesicles by SPR analysis. PKCδ-C2, PKC ε-C2, and PKCη-C2 showed little to no binding. However, PKC θ-C2 exhibited strong binding with a Kd ~ 80 nM. (N. Bhardwaj, R.V. Stahelin, R.E. Langlois, W. Cho, and H. Lu. 2006.J Mol Biol359: 486-495)
Results: Sequence-based Prediction Goal: To identify peripheral proteins on the basis of their sequence-derived features
PU Learning: 2-step strategy • Step 1: Identifying a set of reliable negative examples from the unlabeled set • Spy technique, • 1-DNF technique • Rocchio algorithm. • Step 2: Building a sequence of classifiers by iteratively applying a classification algorithm and then selecting a good classifier • Random Forests • SVM
Step 1 Step 2 positive negative Using P, RN and Q to build the final classifier iteratively or Using only P and RN to build a classifier Reliable Negative (RN) U positive Q =U - RN P This layer is additional to classical ML problems
Step 1 • Sample a certain % of positive examples and put it in unlabeled as “spies”. • Run a classification algorithm assuming all unlabeled examples are negative, • Extract reliable negative examples from the unlabeled set more accurately (threshold).
Step 2: Running the classifier iteratively (1) Running a classification algorithm iteratively • Run SVM iteratively using P, RN and Q until there no example from Q can be classified as negative. RN and Q are updated in each iteration (2) Classifier selection . • 1. Every document in P is assigned the class label 1; • 2. Every document in RN is assigned the class label -1; • 3. i = 1; • 4. Loop • 5. Use P and RN to train a classifier Si; • 6. Classify Q using Si; • Let the set of documents in Q classified as • negative be W; • 8. if W = { } then exit-loop • 9. else Q = Q ∪ W; • 10. RN = RN ∪ W; • 11. i = i +1;
Process with iteration U Q1 Process 1 RN1 Q1 RN2 Q2 Iteration 1 Iteration 2 Process 2: Add spies in every step. U Q1 RN1 Q1 RN2 Q2 (N. Bhardwaj, and H. Lu. in preparation)
Methods: Features used • Overall charge of the domain as electrostatic compatibility is significant (1) • Amino acid composition (20) • Total hydrophobicity (1) • Total α-helix propensity (1) • Total β-sheet propensity (1) • Di-peptide composition (400) (N. Bhardwaj, and H. Lu. in preparation)
Methods: Dataset • Positive • Yeast, Mouse, Human proteomes Manually picked out binding cases (932 cases) reduce seq identity to 40% (232 cases) • Unlabeled • Yeast, Mouse and Human Proteomes extract all domains (32,000 domains) Reduce seq identity to 20% remove binding cases (3,759 cases) • P:U:~1:16 (N. Bhardwaj, and H. Lu. in preparation)
Methods: Evaluation • Removed 40 positive cases for holdout test • Added 15% (28 cases) of positive set to unlabeled set • Implemented the two layers; built the final model • Tested the model on the holdout test • Accuracy and sensitivity (N. Bhardwaj, and H. Lu. in preparation)
Results: Process 1 • With P and U+S as training, identified 877 RN1 examples • Iterated • Converged at 2032 RN cases and 1728 Q cases • Built final model with P and S as positive and RN1 through RN9 as negative 2882 2032 1728 877 • Was tested on the holdout test of 40 proteins • Only two domains were classified as negative (95% accuracy, 95% sensitivity): ARGH2-PH, RASL2-C2 (N. Bhardwaj, and H. Lu. in preparation)
Results: Process 2 • Was tested on the holdout test of 40 proteins • Only two domains were classified as negative (95% accuracy, 95% sensitivity): ARGH2-PH, RASL2-C2 2882 877 (N. Bhardwaj, and H. Lu. in preparation)
Results: Knowledge-Mining Goal: To build classification trees for peripheral proteins while putting forth some biological rules
Introduction • Goal: to identify binding and non-binding cases from the same domain
Knowledge Mining: Features used • Overall charge of the domain as electrostatic compatibility is significant (1) • Amino acid composition (20) • Total hydrophobicity (1) • Total α-helix propensity (1) • Total β-sheet propensity (1) • Local Environment AA composition (80) TYR_YN: Tyr in a high helix and low sheet propensity environment (N. Bhardwaj, and H. Lu. in preparation)
Materials and methods • 232 positive cases from before • 213 negative cases that were manually checked for their function • ADTree was used to build classification tree (N. Bhardwaj, and H. Lu. in preparation)
Tree built on all domains Red: Non-binding Black: Binding (N. Bhardwaj, and H. Lu. in preparation)
C1 domain Red: Non-binding Black: Binding (N. Bhardwaj, and H. Lu. in preparation)