Download
1 / 1
10 likes | 77 Views
This study integrates multiple publicly available gene expression datasets to predict therapeutic options across various diseases. By analyzing data from nearly 70 disease microarray datasets and gene expression data from human cell lines treated with around 160 drugs, potential treatment options are identified. Disease signatures are generated by analyzing upregulated and downregulated genes, followed by standardization and computation of enrichment scores for drug-disease pairs. The results are visualized using a heatmap, demonstrating connectivity scores for drugs and compounds across different diseases. Validation is done through querying known drug-disease associations. This approach enables the discovery of potential functional connections between drugs, genes, and diseases based on common gene expression changes.

E N D
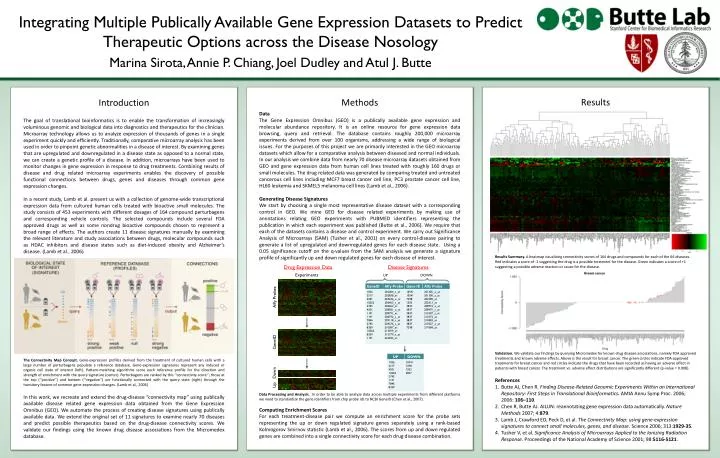
Integrating Multiple Publically Available Gene Expression Datasets to Predict Therapeutic Options across the Disease Nosology Marina Sirota, Annie P. Chiang, Joel Dudley and Atul J. Butte • Results • Results Summary. A heatmap visualizing connectivity scores of 164 drugs and compounds for each of the 66 diseases. Red indicates a score of -1 suggesting the drug is a possible treatment for the disease. Green indicates a score of +1 suggesting a possible adverse reaction or cause for the disease. • Validation. We validate our findings by querying Micromedex for known drug disease associations, namely FDA approved treatments and known adverse effects. Above is the result for breast cancer. The green circles indicate FDA approved treatments for breast cancer and red circles indicate the drugs that have been recorded as having an adverse effect in patients with breast cancer. The treatment vs. adverse effect distributions are significantly different (p-value = 0.008). • References • Butte AJ, Chen R. Finding Disease-Related Genomic Experiments Within an International Repository: First Steps in Translational Bioinformatics.AMIA AnnuSymp Proc. 2006; 2006: 106–110. • Chen R, Butte AJ. AILUN: reannotating gene expression data automatically. Nature Methods 2007; 4:879. • Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006; 313:1929-35. • Tusher V, et al. Significance Analysis of Microarrays Applied to the Ionizing Radiation Response. Proceedings of the National Academy of Science 2001; 98:5116-5121. Methods Data The Gene Expression Omnibus (GEO) is a publically available gene expression and molecular abundance repository. It is an online resource for gene expression data browsing, query and retrieval. The database contains roughly 200,000 microarray experiments derived from over 100 organisms, addressing a wide range of biological issues. For the purposes of this project we are primarily interested in the GEO microarray datasets which allow for a comparative analysis between diseased and normal individuals. In our analysis we combine data from nearly 70 disease microarray datasets obtained from GEO and gene expression data from human cell lines treated with roughly 160 drugs or small molecules. The drug related data was generated by comparing treated and untreated cancerous cell lines including MCF7 breast cancer cell line, PC3 prostate cancer cell line, HL60 leukemia and SKMEL5 melanoma cell lines (Lamb et al., 2006). Generating Disease Signatures We start by choosing a single most representative disease dataset with a corresponding control in GEO. We mine GEO for disease related experiments by making use of annotations relating GEO experiments with PUBMED identifiers representing the publication in which each experiment was published (Butte et al., 2006). We require that each of the datasets contains a disease and control experiment. We carry out Significance Analysis of Microarrays (SAM) (Tusher et al., 2001) on every control-disease pairing to generate a list of upregulated and downregulated genes for each disease state. Using a 0.05 significance cutoff on the q-values from the SAM analysis we generate a signature profile of significantly up and down regulated genes for each disease of interest. Data Processing and Analysis. In order to be able to analyze data across multiple experiments from different platforms we need to standardize the gene identifiers from chip probe ids to NCBI GeneID (Chen et al., 2007). Computing Enrichment Scores For each treatment-disease pair we compute an enrichment score for the probe sets representing the up or down regulated signature genes separately using a rank-based Kolmogorov Smirnov statistic (Lamb et al., 2006). The scores from up and down regulated genes are combined into a single connectivity score for each drug disease combination. Introduction The goal of translational bioinformatics is to enable the transformation of increasingly voluminous genomic and biological data into diagnostics and therapeutics for the clinician. Microarray technology allows us to analyze expression of thousands of genes in a single experiment quickly and efficiently. Traditionally, comparative microarray analysis has been used in order to pinpoint genetic abnormalities in a disease of interest. By examining genes that are upregulated and downregulated in a disease state as opposed to a normal state, we can create a genetic profile of a disease. In addition, microarrays have been used to monitor changes in gene expression in response to drug treatments. Combining results of disease and drug related microarray experiments enables the discovery of possible functional connections between drugs, genes and diseases through common gene expression changes. In a recent study, Lamb et al. present us with a collection of genome-wide transcriptional expression data from cultured human cells treated with bioactive small molecules. The study consists of 453 experiments with different dosages of 164 compound perturbagens and corresponding vehicle controls. The selected compounds include several FDA approved drugs as well as some nondrug bioactive compounds chosen to represent a broad range of effects. The authors create 11 disease signatures manually by examining the relevant literature and study associations between drugs, molecular compounds such as HDAC inhibitors and disease states such as diet-induced obesity and Alzheimer’s disease. (Lamb et al., 2006) The Connectivity Map Concept. Gene-expression profiles derived from the treatment of cultured human cells with a large number of perturbagens populate a reference database. Gene-expression signatures represent any induced or organic cell state of interest (left). Pattern-matching algorithms score each reference profile for the direction and strength of enrichment with the query signature (center). Perturbagens are ranked by this ‘‘connectivity score’’; those at the top (‘‘positive’’) and bottom (‘‘negative’’) are functionally connected with the query state (right) through the transitory feature of common gene-expression changes. (Lamb et al., 2006) In this work, we recreate and extend the drug-disease “connectivity map” using publically available disease related gene expression data obtained from the Gene Expression Omnibus (GEO). We automate the process of creating disease signatures using publically available data. We extend the original set of 11 signatures to examine nearly 70 diseases and predict possible therapeutics based on the drug-disease connectivity scores. We validate our findings using the known drug disease associations from the Micromedex database. Drug Expression Data Disease Signatures Experiments UP DOWN Affy Probes GeneID Up Down






