Download
1 / 1
10 likes | 140 Views
Depleted Uranium induces oxidative stress in isolated kidney mitochondria Fatemeh Shaki 1,2 , Jalal Pourahmad * 1 , Mir-Jamal Hosseini 1 1 Faculty of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran

E N D
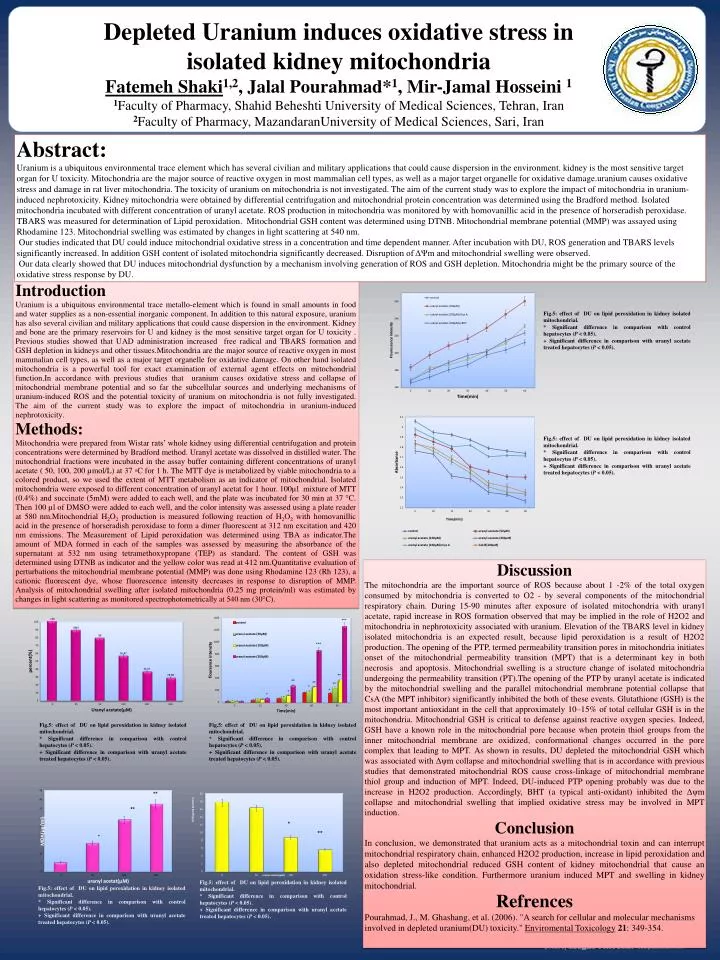
Depleted Uranium induces oxidative stress in isolated kidney mitochondria FatemehShaki1,2, JalalPourahmad*1, Mir-Jamal Hosseini1 1Faculty of Pharmacy, ShahidBeheshti University of Medical Sciences, Tehran, Iran 2Faculty of Pharmacy, MazandaranUniversityof Medical Sciences, Sari, Iran Abstract: Uranium is a ubiquitous environmental trace element which has several civilian and military applications that could cause dispersion in the environment. kidney is the most sensitive target organ for U toxicity. Mitochondria are the major source of reactive oxygen in most mammalian cell types, as well as a major target organelle for oxidative damage.uraniumcauses oxidative stress and damage in rat liver mitochondria. The toxicity of uranium on mitochondria is not investigated. The aim of the current study was to explore the impact of mitochondria in uranium-induced nephrotoxicity.Kidney mitochondria were obtained by differential centrifugation and mitochondrial protein concentration was determined using the Bradford method. Isolated mitochondria incubated with different concentration of uranyl acetate. ROS production in mitochondria was monitored by with homovanillic acid in the presence of horseradish peroxidase. TBARS was measured for determination of Lipid peroxidation. Mitochondrial GSH content was determined using DTNB. Mitochondrial membrane potential (MMP) was assayed using Rhodamine 123. Mitochondrial swelling was estimated by changes in light scattering at 540 nm. Our studies indicated that DU could induce mitochondrial oxidative stress in a concentration and time dependent manner. After incubation with DU, ROS generation and TBARS levels significantly increased. In addition GSH content of isolated mitochondria significantly decreased. Disruption of ΔΨm and mitochondrial swelling were observed. Our data clearly showed that DU induces mitochondrial dysfunction by a mechanism involving generation of ROS and GSH depletion. Mitochondria might be the primary source of the oxidative stress response by DU. Introduction Uranium is a ubiquitous environmental trace metallo-element which is found in small amounts in food and water supplies as a non-essential inorganic component. In addition to this natural exposure, uranium has also several civilian and military applications that could cause dispersion in the environment. Kidney and bone are the primary reservoirs for U and kidney is the most sensitive target organ for U toxicity . Previous studies showed that UAD administration increased free radical and TBARS formation and GSH depletion in kidneys and other tissues.Mitochondriaare the major source of reactive oxygen in most mammalian cell types, as well as a major target organelle for oxidative damage. On other hand isolated mitochondria is a powerful tool for exact examination of external agent effects on mitochondrial function.Inaccordance with previous studies that uranium causes oxidative stress and collapse of mitochondrial membrane potential and so far the subcellular sources and underlying mechanisms of uranium-induced ROS and the potential toxicity of uranium on mitochondria is not fully investigated. The aim of the current study was to explore the impact of mitochondria in uranium-induced nephrotoxicity. Methods: Mitochondria were prepared from Wistar rats’ whole kidney using differential centrifugation and protein concentrations were determined by Bradford method. Uranyl acetate was dissolved in distilled water. The mitochondrial fractions were incubated in the assay buffer containing different concentrations of uranyl acetate ( 50, 100, 200 µmol/L) at 37 ◦C for 1 h. The MTT dye is metabolized by viable mitochondria to a colored product, so we used the extent of MTT metabolism as an indicator of mitochondrial. Isolated mitochondria were exposed to different concentration of uranyl acetatfor 1 hour. 100µl mixture of MTT (0.4%) and succinate (5mM) were added to each well, and the plate was incubated for 30 min at 37 °C. Then 100 µl of DMSO were added to each well, and the color intensity was assessed using a plate reader at 580 nm.MitochondrialH2O2 production is measured following reaction of H2O2 with homovanillic acid in the presence of horseradish peroxidase to form a dimer fluorescent at 312 nm excitation and 420 nm emissions. The Measurement of Lipid peroxidation was determined using TBA as indicator.Theamount of MDA formed in each of the samples was assessed by measuring the absorbance of the supernatant at 532 nm using tetramethoxypropane (TEP) as standard. The content of GSH was determined using DTNB as indicator and the yellow color was read at 412 nm.Quantitativeevaluation of perturbations the mitochondrial membrane potential (MMP) was done using Rhodamine 123 (Rh 123), a cationic fluorescent dye, whose fluorescence intensity decreases in response to disruption of MMP. Analysis of mitochondrial swelling after isolated mitochondria (0.25 mg protein/ml) was estimated by changes in light scattering as monitored spectrophotometrically at 540 nm (30°C). Fig.5: effect of DU on lipid peroxidation in kidney isolated mitochondrial. * Significant difference in comparison with control hepatocytes (P < 0.05). + Significant difference in comparison with uranyl acetate treated hepatocytes (P < 0.05). Fig.5: effect of DU on lipid peroxidation in kidney isolated mitochondrial. * Significant difference in comparison with control hepatocytes (P < 0.05). + Significant difference in comparison with uranyl acetate treated hepatocytes (P < 0.05). Discussion The mitochondria are the important source of ROS because about 1 -2% of the total oxygen consumed by mitochondria is converted to O2 - by several components of the mitochondrial respiratory chain. During 15-90 minutes after exposure of isolated mitochondria with uranyl acetate, rapid increase in ROS formation observed that may be implied in the role of H2O2 and mitochondria in nephrotoxicity associated with uranium. Elevation of the TBARS level in kidney isolated mitochondria is an expected result, because lipid peroxidation is a result of H2O2 production. The opening of the PTP, termed permeability transition pores in mitochondria initiates onset of the mitochondrial permeability transition (MPT) that is a determinant key in both necrosis and apoptosis. Mitochondrial swelling is a structure change of isolated mitochondria undergoing the permeability transition (PT).The opening of the PTP by uranyl acetate is indicated by the mitochondrial swelling and the parallel mitochondrial membrane potential collapse that CsA (the MPT inhibitor) significantly inhibited the both of these events. Glutathione (GSH) is the most important antioxidant in the cell that approximately 10–15% of total cellular GSH is in the mitochondria. Mitochondrial GSH is critical to defense against reactive oxygen species. Indeed, GSH have a known role in the mitochondrial pore because when protein thiol groups from the inner mitochondrial membrane are oxidized, conformational changes occurred in the pore complex that leading to MPT. As shown in results, DU depleted the mitochondrial GSH which was associated with Δψm collapse and mitochondrial swelling that is in accordance with previous studies that demonstrated mitochondrial ROS cause cross-linkage of mitochondrial membrane thiol group and induction of MPT. Indeed, DU-induced PTP opening probably was due to the increase in H2O2 production. Accordingly, BHT (a typical anti-oxidant) inhibited the Δψm collapse and mitochondrial swelling that implied oxidative stress may be involved in MPT induction. Conclusion In conclusion, we demonstrated that uranium acts as a mitochondrial toxin and can interrupt mitochondrial respiratory chain, enhanced H2O2 production, increase in lipid peroxidation and also depleted mitochondrial reduced GSH content of kidney mitochondrial that cause an oxidation stress-like condition. Furthermore uranium induced MPT and swelling in kidney mitochondrial. Refrences Pourahmad, J., M. Ghashang, et al. (2006). "A search for cellular and molecular mechanisms involved in depleted uranium(DU) toxicity." Enviromental Toxicology21: 349-354. Fig.5: effect of DU on lipid peroxidation in kidney isolated mitochondrial. * Significant difference in comparison with control hepatocytes (P < 0.05). + Significant difference in comparison with uranyl acetate treated hepatocytes (P < 0.05). Fig.5: effect of DU on lipid peroxidation in kidney isolated mitochondrial. * Significant difference in comparison with control hepatocytes (P < 0.05). + Significant difference in comparison with uranyl acetate treated hepatocytes (P < 0.05). Fig.5: effect of DU on lipid peroxidation in kidney isolated mitochondrial. * Significant difference in comparison with control hepatocytes (P < 0.05). + Significant difference in comparison with uranyl acetate treated hepatocytes (P < 0.05). Fig.5: effect of DU on lipid peroxidation in kidney isolated mitochondrial. * Significant difference in comparison with control hepatocytes (P < 0.05). + Significant difference in comparison with uranyl acetate treated hepatocytes (P < 0.05).






