Download

1 / 37
370 likes | 580 Views
Heuristic alignment algorithms and cost matrices. Linda Muselaars and Miranda Stobbe. Overview chapter 2. What sorts of alignment should be considered? The scoring system used to rank alignments. The algorithm used to find optimal (or good) scoring alignments.

E N D
Heuristic alignment algorithms and cost matrices Linda Muselaars and Miranda Stobbe
Overview chapter 2 • What sorts of alignment should be considered? • The scoring system used to rank alignments. • The algorithm used to find optimal (or good) scoring alignments. • The statistical methods used to evaluate the significance of an alignment score. Linda Muselaars and Miranda Stobbe
Overview chapter 2 • What sorts of alignment should be considered? • The scoring system usedto rank alignments. • The algorithm used to find optimal (or good) scoring alignments. • The statistical methods used to evaluate the significance of an alignment score. Linda Muselaars and Miranda Stobbe
Contents • Heuristic alignment algorithms • BLAST • FASTA • Linear space methods • Significance of scores • Bayesian approach • Classical approach • Deriving score parameters • PAM matrices • BLOSUM Linda Muselaars and Miranda Stobbe
Contents • Heuristic alignment algorithms • BLAST • FASTA • Linear space methods • Significance of scores • Bayesian approach • Classical approach • Deriving score parameters • PAM matrices • BLOSUM Linda Muselaars and Miranda Stobbe
The term heuristic • A heuristic algorithm is based on empirical information that has no explicit rationalization. • It does not necessarily return the exact answer to the problem under study, but is faster than the algorithm that does and is still very usable. Linda Muselaars and Miranda Stobbe
BLAST • Basic Linear Alignment Search Tool. • Simplification of the Smith-Waterman algorithm. • Uses subsequences of the query sequence to make ‘neighbourhood words’ using a threshold. • When a neighbourhood word matches a subsequence in the database a ‘hit extension’ process is started. Linda Muselaars and Miranda Stobbe
Example Query sequence: q l n f All subsequences: q l, l n, n f Creating neighbourhood words: q l q l, q m, h l, z l l n l n, l b n f n f, a f, n y, d f, q f, e f, g f, h f, k f, s f, t f, b f, z f Linda Muselaars and Miranda Stobbe
FASTA • FAST Alignment. • Fast approximation of the Smith-Waterman algorithm. • Step 1: • Exact short word matches with length ktup • Step 2: • extend to ungapped alignments • Step 3: • identify gapped alignments • Step 4: • dynamic programming restricted to a subregion Linda Muselaars and Miranda Stobbe
BLAST versus FASTA • They both use the same extension method. • They both can be used for both DNA and proteins. • BLAST is faster than FASTA. • BLAST is more sensitive than FASTA on proteins. • BLAST is less sensitive than FASTA for nucleic acid sequences. • BLAST uses neighbourhood words, FASTA does not. • BLAST is mainly for ungapped alignment, FASTA for gapped alignments. Linda Muselaars and Miranda Stobbe
BLAST vs. FASTA, example • Consider the sequences: n f l and n y l • ktup = 2 (remember: only for FASTA) • Even though FASTA only needs a matching word of size 2 it does not find a match. • BLAST does find a match (of word size 3 even) on account of neighbourhood words. Linda Muselaars and Miranda Stobbe
Demo at www.ebi.ac.uk Linda Muselaars and Miranda Stobbe
Contents • Heuristic alignment algorithms • BLAST • FASTA • Linear space methods • Significance of scores • Bayesian approach • Classical approach • Deriving score parameters • PAM matrices • BLOSUM Linda Muselaars and Miranda Stobbe
Reducing memory usage • Score matrices so far are of size nm (with n and m the sequence lengths). • We can reduce memory usage to n+m. • Cost: time is doubled. • This is done by linear space methods. Linda Muselaars and Miranda Stobbe
Divide and conquer • We find a cell (u,v) in the middle column that is on the optimal path. • This cell divides the matrix in four parts of which two are important for the path. • This is done recursively to these two parts. Linda Muselaars and Miranda Stobbe
Contents • Heuristic alignment algorithms • BLAST • FASTA • Linear space methods • Significance of scores • Bayesian approach • Classical approach • Deriving score parameters • PAM matrices • BLOSUM Linda Muselaars and Miranda Stobbe
Short review • Letter a occurs independently with frequency qa in the random model. • Aligned pairs of residues occur with a joint probability pabin the match model. • Random model: P(x,y|R) = ΠkqxkΠlqyl • Match model: P(x,y|M) = Πkpxkyk Linda Muselaars and Miranda Stobbe
Bayesian approach: model comparison Linda Muselaars and Miranda Stobbe
Comparison • For global matches compare with 0 to determine whether the alignment is significant. • When setting the prior odds ratio in inverse proportion to the size of the database N, compare with log N. • For local matches compare with 0.1 • log(nm) Linda Muselaars and Miranda Stobbe
Extreme value distribution • Scores of a sequence aligned to a set of random sequences obey EVD. • We compute the probability that the best match of unrelated sequences has score greater than our maximal score. Linda Muselaars and Miranda Stobbe
Other alignments • For local ungapped alignments we have a different EVD than for fixed ungapped alignment (because we have more possible starting points). • For gapped alignments empirically established distributions are used. Linda Muselaars and Miranda Stobbe
Correcting for length • When database sequences are longer, we have higher scores. • Solutions: • Subtract log (mi) for length mi of the database sequence. • Bin all the database entries by length and fit a linear function. Linda Muselaars and Miranda Stobbe
Notes on test statistic • Search statistic is the same as the test statistic. • Advantage: both have highly discriminative power. • Disadvantage: introduction of bias in test phase. Linda Muselaars and Miranda Stobbe
Contents • Heuristic alignment algorithms • BLAST • FASTA • Linear space methods • Significance of scores • Bayesian approach • Classical approach • Deriving score parameters • PAM matrices • BLOSUM Linda Muselaars and Miranda Stobbe
Substitution and gap scores • Letter a occurs independently with frequency qa in the random model. • Aligned pairs of residues occur with a joint probability pabin the match model. • f(g) is a function of the length of the gap Linda Muselaars and Miranda Stobbe
Estimating probabilities • Simple approach: set the probabilities to normalised frequencies (assessed by counting frequences in confirmed alignments). • But: • It is difficult to obtain a good random sample. • Does not take into account different ‘distances’ to the common ancestor. Linda Muselaars and Miranda Stobbe
PAM matrices • Percentage of Acceptable point Mutations per 108 years matrices. • Amino acid substitution matrices. • Obtain substitution data from alignments and estimate probabilities for longer evolutionary distances. • A PAMn: n accepted mutations event per 100 amino acids. Linda Muselaars and Miranda Stobbe
PAM matrices (2) • Construct phylogenetic trees relating the sequences in 71 families (at least 85% similar). • Count the number of amino acid changes with respect to immediate ancestor. • 20 x 20 amino acid substitution matrix computed. • Expected number of substitutions is 1% in PAM1. • PAMn = (PAM1)n. • PAM-matrix converted to a log-odds matrix. Linda Muselaars and Miranda Stobbe
Drawbacks • Using the matrix for short time intervals to compute the ones for longer time intervals does not capture the true difference. • Takes into account only single base changes instead of all types of codon changes. • Databases containing alignments of more distantly related proteins are used to derive matrix scores more directly and accurately. Linda Muselaars and Miranda Stobbe
BLOCKS database • Used to derive BLOSUM matrices. • Sequences are clustered according to percentage of identical residues. • Aab then is the frequency of observing a in one cluster aligned to b in another cluster. • Size of the clusters needs to be corrected for. Linda Muselaars and Miranda Stobbe
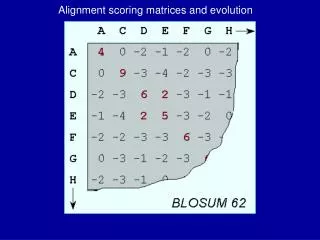
BLOSUM • BLOcks SUbstitution Matrix • BLOSUMn is the matrix where two sequences are put into one cluster when more then n% of their residues are identical (lower n corresponds to longer evolutionary time). • From Aabqa and pab are estimated, which are used to compute the scores for the matrix. Linda Muselaars and Miranda Stobbe
Based on global alignments. PAM1 is the matrix calculated from comparisons of substitutions in unit time. Other PAM matrices are extrapolated from PAM1. Based on local alignments. BLOSUMn is a matrix calculated from sequences with no less than n% divergence. All matrices are based on observed alignments. PAM versus BLOSUM Linda Muselaars and Miranda Stobbe
Gap penalties • Time-dependent: • Number of gaps increases (gap-open score d linear in log t). • Length distribution constant (gap-extend score e remains constant). • In practice people choose gap costs empirically (only two parameters). • As gaps become more likely we could reduce the pairwise scores. Linda Muselaars and Miranda Stobbe
Notes • Objective was to determine whether two sequences are related. • Scoring schemes and statistics to determine the significance of a match. • Even so, it is not always possible to distinguish between two related sequences or two sequences that seem to be related, but are not. Linda Muselaars and Miranda Stobbe
Summary • BLAST and FASTA packages are used to reduce the time used for finding alignments. • Linear space alignments can be used to reduce memory usage. • We need the significance of scores for the importance of a match. • We can use the score parameters stated in PAM and BLOSUM matrices. Linda Muselaars and Miranda Stobbe