Download

1 / 51
530 likes | 1.19k Views
Signal Reception: G Protein-Coupled Receptors. Neurotransmitter receptors. Ligand – gated channels: Nicotinic acetylcholine receptor NMDA-type glutamate receptor Glycine receptor GABA A receptor Serotonin receptor (5-HT 3 ) G protein-coupled receptors:

E N D
Neurotransmitter receptors • Ligand – gated channels: • Nicotinic acetylcholine receptor • NMDA-type glutamate receptor • Glycine receptor • GABAA receptor • Serotonin receptor (5-HT3) • G protein-coupled receptors: • Muscarinic acetylcholine receptor (several types) • Catecholamine receptors • Histamine receptors (H1, H2) • 5-HT receptors other than 5-HT3 • GABAB receptors • ‘Metabotropic’ glutamate receptors • Peptide receptors (Endorphin, cholecystokinin..)
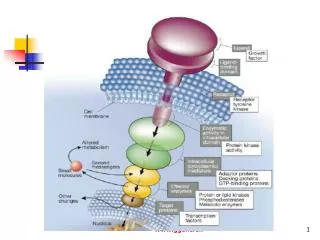
The G Protein-Coupled Receptor (GPCR) Superfamily • Largest known receptor family – Constitutes > 1% of the human genome. • Comprises receptors for a diverse array of molecules: neurotransmitters, odorants, lipids, neuropeptides, large glycoprotein hormones. • Odorant receptor family alone contains hundreds of genes. • Mammalian GPCRs: nearly 300 different kinds – grouped into 3 main subfamilies:
Three Main Mammalian GPCR Subfamilies • Rhodopsin-like group – includes most of the GPCRs. • Glucagon-like group. • Metabotropic glutamate (mGlu) and GABABreceptor family.
Three Main Mammalian GPCR Subfamilies (cont’d) • Grouped according to > 20 % sequence homology. • Databases for the classification of receptors into subfamilies, phylogenetic trees, chromosome localization, ligand binding constants and receptor mutations can be found atwww.gpcr.org/7tm
Almost all Receptors Comprise a Number of Subtypes • Dopamine receptors - 5 subtypes • 5-HT receptors – 13 subtypes • mGlu receptors - 8 subtypes • Acetylcholine receptors – 5 subtypes • Identified by their pharmacological and functional characteristics, rather than by strict sequence homology: - Some receptors for the same ligand show remarkably little homology (e.g., histamine H3 and H4 have the lowest recorded homology (~ 20 %) to other histamine receptors H1 and H2).
Each GPCR family contains some orphan receptors, which have been identified as members of the GPCR superfamily by homology cloning but whose activating ligand is unknown. • But high throughput screening has recently added to the advances in being able to identify the ligand.
Originally published in Science Express on 25 October 2007. Paper version: Science 23 November 2007: Vol. 318. no. 5854, pp. 1258 - 1265. High-Resolution Crystal Structure of an Engineered Human β2-Adrenergic G Protein–Coupled Receptor VadimCherezov, Daniel M. Rosenbaum, Michael A. Hanson, Søren G. F. Rasmussen, Foon Sun Thian, Tong Sun Kobilka, Hee-Jung Choi, Peter Kuhn, William I. Weis, Brian K. Kobilka,Raymond C. Stevens Heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors constitute the largest family of eukaryotic signal transduction proteins that communicate across the membrane. We report the crystal structure of a human β2-adrenergic receptor–T4 lysozyme fusion protein bound to the partial inverse agonist carazolol at 2.4 angstrom resolution. The structure provides a high-resolution view of a human G protein–coupled receptor bound to a diffusible ligand. Ligand-binding site accessibility is enabled by the second extracellular loop, which is held out of the binding cavity by a pair of closely spaced disulfide bridges and a short helical segment within the loop. Cholesterol, a necessary component for crystallization, mediates an intriguing parallel association of receptor molecules in the crystal lattice. Although the location of carazolol in the β2-adrenergic receptor is very similar to that of retinal in rhodopsin, structural differences in the ligand-binding site and other regions highlight the challenges in using rhodopsin as a template model for this large receptor family.
α, β, γ Subunits α Subunit (23 isoforms): contains the GTP/GDP binding site is responsible for identity. β (5 isoforms) and γ (12 isoforms): are identical or very similar, interchangeable in vitro; most of them are ubiquitously expressed; membrane anchored through prenylation of Gβ. Golf: expressed in olfactory bulb, coupled to PLCβ. GT (transducin): is coupled to cGMPphosphodiesterase and is expressed in the rod cells of the retina (these cells are Inactivated by light!!): hν hits rhodopsin -> opsin is activated -> facilitates GTP loading of GT -> activates cGMPphosphodiesterase -> cGMP (keeps Na+ and Ca2+ channels open to cause depol -> nt release) -> converted to 5’GMP (inactive => channels closed => membrane polarization => no nt released).
Receptor Family 1 – Rhodopsin Family N Y Y C C Extracellular TM1 TM2 TM3 TN4 TM5 TM6 TM7 Lipid Bilayer CC D R Y Intracellular C
Receptor-Ligand Interactions:Rhodopsin-like Family 6 5 S OH 7 N F OH S HO +N 1 4 D 3 2
Receptor Family 2 – Glucagon-like Family • Structurally similar to that of Family 1, except that they have a much larger N-terminal domain, which contains multiple potential S-S bridges. • Most of the ligands binding to these receptors are peptides or glycoprotein hormones: - 30-40 residues is typical. - likely to interact with the receptor over large surface areas.
Receptor Family 2 – Glucagon-like Family N Y Y C C Extracellular TM1 TM2 TM3 TN4 TM5 TM6 TM7 Lipid Bilayer CC D R Y Intracellular C
Receptor Family 3 – mGluR/GABAB Family • Extremely large extracellular N-terminal ligand binding domain. • Highly conserved 3rd short intracellular loop. • Shares only ~ 12 % sequence homology with that of Family 1, but the overall transmembrane topology is similar. • Forms dimers: - GABAB receptor forms heterodimers between GABABR1 and GABABR2 through coiled coil regions in the C-terminal tails. - This dimerization is required for efficient cell surface expression and signalling. - Metabotropic glutamate receptors dimerization is stabelized by disulfide bonds in the N-terminal extracellular domain.
D D D Dissociation of receptor-G protein complex Drug binding and G protein activation αα αα βα βα GDP GTP γ γ αα βα γ GTP Inactivation of Gα through intrinsic GTPase activity Reformation of receptor G protein complex Pi αα The G Protein Cycle GDP
Receptor-G protein InteractionsHow are receptor-G protein interactions measured? Low- affinity R + G(GTP-δ-S) RG(GDP) GTPγS GDP Without GTP, both high- and low-affinity states are measured. With GTP and Mg2+, only low-affinity state is measured, because Agonist binding rapidly induces change from high- to low-affinity. Ligand-binding assays: High-affinity
Receptor-G protein Interactions Structural features of receptors involved in G protein activation How does agonist binding cause receptor conformational change? • Agonists vary in their binding affinity for the GPCR = drug-receptor interaction. • How well the drug causes a conformational change in the receptor to activate G proteins = efficacy. • There are multiple agonists (partial, full) with different binding affinities.
Receptor-G protein Interactions Structural features of receptors involved in G protein activation • Mechanism of conformational change highly conserved. • Constraining intermolecular interactions that keep receptors preferentially silent in the absence of agonist: such as between TM5 & TM6 and between TM3 & TM7. • E.g., ‘DRY’ motif in TM3 (earlier). • Upon receptor activation, the arg is protonated adjacent residues move tilting the TM helix incr exposes previously hidden sequences, which interact with G protein. • Much evidence for preceding. • However, the exact aa sequence responsible for this has been difficult to pinpoint.
Constitutive Activity • Many receptors show constitutive activity even when expressed at physiol levels (e.g., rat dopamine D1, rat, human hist H2, human dopamine D3, and human 5-HT1A). • Inverse agonists. • Mutations have been identified that incr the basal activity w/o affecting the ability of agonists to further activate the receptors. • These mutations affect stabilizing interactions between helices that hold the receptor in an inactive state and those interfering with these interactions
Multiple active conformations – stimulus trafficking • There may be several active conformations, which are induced by certain drugs = stimulus trafficking. • Different drugs can promote distinct receptor conformations, which interact with different G proteins resulting in activation of distinct signaling pathways. • E.g., partial agonists at human 5HT2A and 5HT2C receptors differential stimulation of IP and AA 2nd messenger signaling systems.
Cell-type Specific Factors • Receptor Splice Variants • Levels of receptor expression and signal amplification (see next slide). - with high receptor density + strong coupling to G protein pathway, the [drug] required to generate 2nd messengers may << [drug] required to occupy a significant fraction of receptors. - This system will show a large amount of signal amplification. - ‘receptor reserve’ = ‘spare receptors’ = ‘strong coupling’. - Signal amplification is fast. - Equilibrium is reached quickly – depends on the rate constants for association and dissociation.
Signal Amplification and Receptor Reserve Response 100 80 60 40 20 10 KD = 100 nM– good enough in a strongly coupled system (left shift). In contrast, the same receptors in this cell may also signal through another, less well coupled pathway with less signal amplification and less receptor reserve. Response Binding % of Max 0.01 0.1 1 10 100 1000 10000 Drug (nM)
E (Ca2+ channels) G (α01/β3/γ) G (α02/β1/γ) Specificity of receptors for G protein subtypes Some receptors can show selectivity for a certain α subtype in 1 type of cell, but not in another cell type: R (muscarinic) R (somatostatin)
Restricted localization GPCRs undergo the same trafficking we have discussed earlier (Protein trafficking and LGIC slides).
Regulation of G protein-coupled receptor function Desensitization/resensitization– a decrease in responsiveness during continuous drug application or a right-shift in a drug dose-response curve. After removal of the drug, receptor activity recovers, although the speed and extent of this resensitization can depend on the duration of agonist activation. Rapid desensitization (sec-min) results from receptor phos, arrestin binding, and receptor internalization. Long-term desensitization (down-regulation) involve changes in receptor and/or G protein levels, and their mRNA stability and expression. Long-term changes in [GPCR]s and [accessory proteins]s known to be induced by chronic drug treatment and involved in several pathologies.
Phosphorylation 2nd messenger kinase G protein receptor kinase (GRK) Arrestin β-arrestin binding to phosphorylated GPCR is required to decrease GTPase activity prior to desensitization. Receptor trafficking, internalization, and recycling (covered earlier; see Protein trafficking and LGIC slides).
Mechanisms of long-term down regulation Long-term (> 1 hr) treatment with agonist induces the loss of total cellular receptor number in addition to the decr in surface receptor number. e.g., antidepressants (e.g., fluoxetine) incr [5HT]synapse decr 5HT receptor density. Receptor endocytosis: C-terminal domain determines whether they enter the recycle pathway or the lysosomal pathway: - 2 distinct motifs: 1. PDZ-domain interats with NHERF in a phos-dependent manner. 2. A short sequence that interacts with NSF (N-ethylmaleimide sensitive factor). Arrestin has also been shown to be important for recycling: e.g., V2 vasopressin receptor, which continues to bind arrestin while in endosomes, does not recycle back to plasma membrane.
Regulation at the G protein level Regulator of G protein signaling (RGS = GAPs = GTPase activating proteins) family of proteins (> 20 members) regulate the rate of GTP hydrolysis in the Gα subunit. Can also attenuate G protein actions that are mediated by βγ subunits, because they can alter the number of βγ available by enhancing the affinity of Gα subunits for the βγ after GTP hydrolysis incr rate of reformation of the heterotimer.
Regulation at the G protein level (cont’d) RGS proteins also important in regulating the temporal characteristics of G protein actions. E.g., RGS proteins accelerate the decay of agonist-induced activation of GIRK (G protein regulated inward rectifying K channels). E.g., RGS proteins accelerate desensitization of adrenergic receptor-induced N-type Ca2+ channel currents.
Mechanisms of Receptor Regulation D D D D P P P P P P Arrestin Arrestin αα (2) Phosphorylation Clathrin βα (3) Arrestin binding Clustering in clathrin-coated pits γ (7) Recycling Agonist binding and G protein activation (5) Endocytosis Endosomes D P P • Dissociation of agonist: • Dephosphorylation • Sorting between cycling • and lysosomal pathways Arrestin (8) Traffic to lysosomes Lysosomes
Expanding roles forβ-arrestins as scaffolds and adaptors in GPCR signaling and trafficking.(Miller & Lefkowitz, 2001. Curr. Opin. Cell Biol. 13:139-145).
N g b a How G-protein-coupled receptors work (1) extracellular space ‘7TM’ - receptor cytosol a g b heterotrimeric G-protein Ligand GDP GTP GDP
N N a GTP g b How G-protein-coupled receptors work (2) active P inactive a g b GDP
g b a GTP How G-protein-coupled receptors work (3) Adenylate cyclase active inactive ATP cAMP cAMP active inactive Protein kinase A Phosphorylation of multiple target proteins
as GTP ai GTP Some G-proteins are inhibitory b-Adrenoceptor AC active a2-Adrenoceptor AC inactive
g b K+ ai GTP bg-Subunits of G proteins may have regulatory activity, too Muscarinic (M2) acetylcholine receptor Kir AC inactive
as adenylate cyclase cAMP protein kinase A ai1 adenylate cyclase cAMP protein kinase A Ga-proteins regulate diverse effector systems at cGMP phosphodiesterase cGMP aq phospholipase C protein kinase C PIP2 IP3 + DAG Ca++ phosphorylation of multiple proteins ER
Many transmitters have multiple GPCR with different downstream signaling mechanisms Norepinephrine, a1 IP3 + DAG epinephrinea2 cAMP b1,b2 cAMP Dopamine D2 - D4 cAMP D1, D5 cAMP Acetylcholine M1,,M4,M5 IP3 + DAG M2, M3 cAMP
Cycloexhyladeonsine binding (relative) log [PD 81,723] ‘Allosteric’ agonists PD 81,723
Cooperative binding to GPCR oligomers may explain the behaviour of pseudo-allosteric agonists
Agonist-specific coupling of a1-adrenergic receptors Arachidonic acid (% basal) Inositol-P (% basal) Log [agonist] (M) Efficacy: NA = pOCT > mOCT NA > mOCT > pOCT
Coupling to multiple G-proteins in the two-state model Antagonist Agonist GPCR active GPCR inactive G-protein A, Effect A G-protein B, Effect B