Download

1 / 37
430 likes | 744 Views
3. Atomic structure and atomic spectra. 3.1. Structure and spectra of hydrogenic atoms 3.1.1 The structure of hydrogenic atoms 3.1.2 Atomic orbitals and their energies 3.1.3 Spectroscopic transitions and selection rules 3.2. The structure of many electron atoms

E N D
3. Atomic structure and atomic spectra 3.1. Structure and spectra of hydrogenic atoms 3.1.1 The structure of hydrogenic atoms 3.1.2 Atomic orbitals and their energies 3.1.3 Spectroscopic transitions and selection rules 3.2. The structure of many electron atoms 3.2.1 The orbital approximation 3.2.2 The spectra of complex atoms 3.2.3 Singlet and triplet states H Xe
3.1 Structure and spectra of hydrogenic atoms Hydrogenic atoms: atoms having only one electron (H, He+, Li2+, …) → the Schrödinger equation can be solved Many-electron atoms→impossible to solve the Schrödinger equation→ approximations are needed to solve the SE (Hartree-Fock Theory, Density Functional Theory, etc..). 3.1.1 The structure of hydrogenic atoms Rydberg observed that the lines in the emission spectrum of H fit the expression: where H is the Rydberg constant for H (H= 109677cm-1). One series is characterized by n1, within this series the lines are specified with n2 that can take the values n1+1, n1+2, … For n1=1 →Lyman series in the ultraviolet region For n1=2 →Balmer series in the visible region For n1=3 →Paschen series in the infrared region
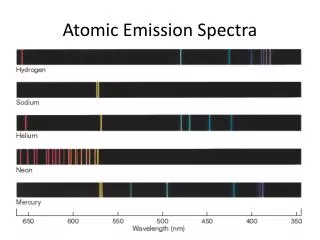
Hydrogen spectrum The gas inside the tube; that emits blue light is mercury, but the same can be done with H2. The gas pressure in the tubes are low, because of natural radioactivity and cosmic rays there are always few free electrons and ions. When something around 2000-3000 volts is applied to these tubes under low pressure, free electrons and ions are accelerated and collide with the gas molecule H2 and dissociate them in excited Hatoms. These collisions generate an avalanche of charged particles. As a result, the gas in the discharge tube contains a lot of ionized and highly excited atoms. The excited atoms give back this energy in the form of light, while they are turning back to their ground states. (NB: Neon tubes are typical examples of discharge tubes) The electromagnetic radiation emitted is then analyzed by a spectrometer and various emission lines of hydrogen can be observed in the visible (Balmer series), the ultraviolet (Lyman series) and infra-red (Paschen series).
From the experimental law by Rydberg, a general principle is found: the Ritz combination principle. It states that the wavenumber of any spectral line is the difference between two terms. NB: it’s only for hydrogenic atoms that the terms have the form: Bohr frequency condition: when an atom changes its energy by E, the difference is carried away as a photon of frequency : E = h In the emission spectrum of an atom, a line appearing at a frequency corresponds to a change in energy E=E2-E1= hc (T1-T2) of an atom that is characterized by the difference between two terms T. The atom can only have specific energies: quantization. What is the origin of these permitted discrete values of energy?
A. The Schrödinger equation for the electron of a hydrogenic atom Atom = 1e- + 1 nucleus of mass mN. Both move and have a kinetic energy Te and TN. Because of the electron charge and the positive charge |Ze| of the nucleus, the Coulomb potential energy V(r) has to be taken into account in the energetics of the system. The Hamiltonian of the Schrödinger equation H tot = E totdescribing the system is: B. Separation of internal motions The atom is a 2-body system: both the nucleus and e- move with respect to each other (relative motion) and both move with respect to an outside reference point. However, it’s possible to separate the relative motion of two particles from the motion of the center of mass. Moreover, since me<< mN, we’ll show that the simplified picture of an electron moving in a spherical potential created by the nucleus can be envisaged.
In classical mechanics: let’s try to express total energy of two bodies (electron and nucleus) not with respect to their coordinates x1 and x2, but with respect to the position of the center of mass X (of the atom) and the distance x between the two bodies (i.e; the distance between the electron and the nucleus). m= total mass of the system P= linear momentum of the system as a whole = reduced mass p= “internal” linear momentum NB: This way to analyze the motion of a multi-body system will be used in Chap 16
Via the correspondence principles, the Hamiltonian of the system (hydrogenic atom) can then be expressed in 3D with respect to the coordinates of the center of mass (c.m.) and the relative coordinate (distance between the e- and the nucleus). The total wavefunction tot appears as the product of a wavefunction c.m.describing the motion of the center of mass of the system (e- + N) and an electronic wavefunction characterizing the motion of the e- around the nucleus: tot = c.m. The total Schrödinger equation is then split in two equations: (1) H tot = Etot tot c.m. =tot (2) NB1: Concerning equation (1), we concluded in chap 2 that the translational energy of an atom in gas phase is not quantized and can be described in classical physics. We now focus on Equation (2)
z r y x C. Separation of the variables in the electronic wave function In Equation (2), ≃mebecause mN>>me Equation (2) describes the electron moving in the spherical potential of the nucleus. It should be solved to rationalize the presence of lines in the emission spectrum of the atoms. In the rest of this chapter, we’ll only consider this electronic Schrödinger equation: From chap 2 (rotation in 3D), the SE in spherical coordinates is: Since the potential is spherical (does not depend of angles, and ): ×(r2/RY) C-C
NB: For the separation of the SE equation C is arbitrary. However, the angular equation is an eigenvalue equation, which has the eigenvalues: -l(l+1). Hence, for the sake of simplicity in the mathematical development C is chosen to be directly: (1’) and (2’) As seen in Chap 2, the solutions Y(,) of (2’) are the spherical harmonics that are specified by the quantum numbers l and ml. Let’s focus now on the radial wave equation (1’) that describes the motion of a particle of masse in a one-dimensional region where the potential is Veff(r). D. The radial solutions R(r) Coulombic potential created by the nucleus Potential coming from the centrifugal force that arises from the angular momentum of the e- turning around the nucleus For l=0: e- attracted by the nucleus; ⍱r (purely Coulombic) For l≠0:e- has an angular momentum that gives a positive contribution to Veff→ repulsion from the nucleus.
Solving the radial equation (1’) gives eigenvalues En characterized by a quantum number: n= 1, 2, 3, … The solutions of (1’) are the Hydrogenic radial wavefunctions Rn,l(r), which are specified by 2 quantum numbers n and l. They have the form: L= polynomial in : the associated Laguerre polynomial. (no dimension) Bohr radius = radius of the electron orbit of the lowest energy; a0= 0.529 Å The general form of these solutions are submitted to 2 constrains: (i) At the nucleus (i.e.; r=0 or =0), the solution should never be infinite: R(0) ≠∞, because the probability density cannot be infinite. (ii) Far away from the nucleus (i.e.; r →∞): R(∞) ≠∞ In order to fulfill those conditions, the values of n and l must respect that criterion: l ≤ (n-1) and n=1, 2, 3, ...
For l≠0: R(0)= 0, the wavefunction is zero at the nucleus because of the centrifugal force pushes away the electron For l= 0: R(0)= a finite value, because the attractive Coulombic potential localizes the electron close to the nucleus l=0 l=1 l=2 l=0 l=1 l=2
3.1.2 Atomic orbitals and their energies Associated Laguerre polynomials multiplied by an exponential, and characterized by n and l Atomic orbital is a one-electron wavefunction defined by 3 quantum numbers (n, l, ml) Spherical harmonics specified with l and ml = × Principal quantum number: n= 1, 2, 3, … In an orbital characterized by n, the energy of the electron is : Angular momentum quantum number: l = 0, 1, 2, …, n-1 In an orbital characterized by l, the angular momentum due to the rotation of the electron around the nucleus is: Magnetic quantum number: ml= l , l-1, l-2 , … , - l In an orbital characterized by ml, the plane of rotation of the electron has a specific orientation characterized by the z-component of L : NB: A spin function should be added in the wavefunction. Hence, a spin quantum number,ms = ∓½, should also be specified in order to characterize totally the electron.
A. The energy levels and the ionization energies The energies given by the formula are negative, they correspond to a bound state of the electron. The zero energy, reference level for the energy, corresponds to a situation where the electron is not bound to the proton H+ and has a zero kinetic energy. It is characterized by n = ∞ The positive energies correspond to unbound states of the electron. The wavefunction describing such an electron is also solution of the SE, and it is the wavefunction of a free electron. The energy of the unbound electron are not quantized and form the continuum states of the atom. The ionization energy, I, of an atom is the minimum energy required to remove an electron from the atom that is in its ground state (the state of lowest energy of the atom). e.g: For H, there is one electron. The atom is in its ground state when the electron is in the state characterized by n=1. I=-En=1= hc H= 2.179 10-18J= 13.6 eV
B. Shells and subshells In hydrogenic atoms, all orbitals in the same shell have the same energy (characterized by n). NB: In many-electron atoms, we’ll see that the subshells have different energies. ml≤ |l | ml l Number of orbitals with different ml l ≤ (n-1) n = 1 2 3 4 K L M N l = 0 1 2 3 s p d f n
C. s orbitals For Hydrogen (Z=1), the 1s orbital (n= 1, l= 0, ml= 0) has spherical symmetry. The wavefunction decays exponentially from a maximum value, 1/(a03)1/2, at the nucleus. T<<, V>> <T> = -1/2 <V> Radial node Actual ground state wave-function has to fulfill the Virial theorem 2s 1s Virial theorem: for V(r)= rb→ 2 <T> = b <V>. For n=1, l=0, V(r) is Coulombic= r -1 →<T> = -1/2 <V>. T>>, V<< All the s orbitals are spherical, but differ by the number of radial nodes (where the probability to find the electron at the node is zero). The higher the “n”, the higher the number of nodes. Because: the higher the “n”, the more there are solutions for the associated Laguerre polynomial Ln,l. The position rnode of the nodes are found by solving Ln,l= 0, such that R(rnode)= 0 →2(rnode)=0. High curvature→ T >> and V << High amplitude→ e- is close to the nucleus
D. The mean radius <r> This is the expectation value of the operator “r” Spherical coordinates For a given n, the mean radius follows the order d < p < s
E. Radial distribution functions For a s orbital, the probability density ||2to find the electron is higher close to the nucleus: ||2 ∝ exp(-2Zr/a0) But, close to the nucleus, there is not a lot of room for the electron to be. Hence, the probability to find the electron in a spherical shell of thickness dr is lower. Because the volume of the shell dτ= 4r2 dr (surface of a sphere × thickness) is smaller. The most probable radius at which the electron will be found is a compromise between the volume of the spherical shell and the probability density ||2. The probability P(r)dr of finding the electron at any angle (∫∫dd) at a constant radius r is the integral of the probability density ||2 over the surface of a sphere of radius r: For the 1s orbital of H, the maximum of P(r) occurs at r=a0, the Bohr radius.
http://www3.adnc.com/~topquark/quantum/hydrogenmain.html For the stationary states nlm(x,y,z) of the hydrogen atom, the probability densities |nlm(x,0,z)|2 are plotted in a plane containing the z axis.
spherical coordinates: x= r sincos; y= r sin sin; z= r cos z r y x F. p orbitals Different rotations: clockwise, counter-clockwise These linear combinations are real functions. They are also solutions of the SE for the atom, but have no net angular momentumvs. the z-axis compared to p1. - + Nodal plane
Demonstration: “if 1 and 2 are solutions of the Schrödinger Equation, i.e., H1= E 1 and H2= E 2, then the linear combination = c11+ c22 is also a solution” H = H (c11+ c22)= c1 H1+ c2 H2 H = c1 E1+ c2 E2= E(c11+ c22) H = E Hence, is indeed also a solution. It means that px and py are solutions of the SE for a hydrogenic atom
F. d orbitals In the shell n=3, there are 3 subshells: → l= 0→ ml= 0 → 1 s-orbital → l= 1→ ml= -1, 0, 1 → 3 p-orbitals → l= 2→ ml= -2, -1, 0, 1, 2 →5 d-orbitals From a linear combination of these orbitals, real orbitals can be built. They are also solutions of the SE for the hydrogenic atom: dxy, dyz, dzx, dx2-y2, dz2
http://www.albany.net/~cprimus/orb/ The electron orbitals presented here represent a volume of space within which an electron would have a certain probability of being based on particular energy states and atoms.
3.1.3 Spectroscopic transitions and selection rules From the Bohr frequency condition, the excess of energy produced when the atom undergoes a transition from an excited state (characterized by an electron in the orbital n1,l1,ml1) to a lower energy state (for which the electron is in the orbital n2,l2,ml2) is discarded as a photon of frequency . All the possible transitions, (n1,l1,ml1) →[(n2,l2,ml2)+h], are not permissible because the global angular momentum of the system should be conserved. The photon has an intrinsic spin angular momentum corresponding to s=1. The change in angular momentum of the electron must compensate for the angular momentum carried away by the photon. d orbital (l=2) → s orbital (l=0) + h (s=1) Transition forbidden because 2≠ 0 + 1 The probability for a transition to occur, i.e. the intensity of the different lines in the spectrum, is given by the transition dipole momentfibetween an initial state i and a final state f :
The use of transition dipole moment to evaluate the intensity of a line is a general method that can be applied to any atoms or molecules. For a hydrogenic atom, the introduction of the final and initial states, characterized by two different sets of numbers (n, l, ml), leads to the selection rules indicating which transition is allowed: l= ±1 ml= 0, ±1 Note: There is no specific rule for the principal quantum number, because it does not carry information about the angular momentum.
Absorption spectrum of Saturnus The light emitted from the sun is absorbed by atoms contained in the ring of the planet. The absorption spectrum gives us the composition of the ring: ice, mineral, iron ... The shift in frequency observed for an absorption line appearing on both sides of the ring is explained like this: the ring is composed of particles (from 10 cm to 10 m) rotating around the planet at a high velocities. The spectral line appears shifted from the earth because of the speed of rotation of the particles: the Doppler effect modifies the apparent frequency of the line. From the measured frequency shift, the speed of the particles can be deduced (20km/s).
3.2 Structure of many-electron atoms The Schrödinger equation for a three-body system cannot be solved analytically (e.g.: He= 1 nucleus, 2 electrons) → approximations required. 3.2.1 The orbital approximation The spatial electronic wavefunction of an atom with n electrons is written: (r1, r2,..., rn), where the ri are the vectors between the nucleus and the electron i. Because the electrons interact with each other via a Coulombic potential Vee, (r1, r2,..., rn) is exactly unknown. The electronic Hamiltonian of the atom is: A first approximation is to neglect the interaction between electrons. Consequently, the Hamiltonian can be divided in a sum of terms, each acting on one electron: the monoelectronic Hamiltonians hi. The Schrödinger equation can be split in n monoelectronic equations: hi (ri)=i (ri) i=1, … n The wavefunction of the atom is then the product of monoelectronic wavefunctions (ri). This is the orbital approximation: each electron occupies its “own” orbital (ri) : (r1, r2,..., rn)= (r1) (r2) ... (rn)
In order to associate an orbital (ri) to each electron in a many-electron atom, we can think to use the solutions of the SE for a hydrogenic atom (1e- + 1 nucleus of charge Z). He: 2e- + nucleus (Z=2) →electron configuration: 1s2….but is it ↓↑ or ↑↑ ? The answer is given by the Pauli exclusion principle. Li: 3e- + nucleus (Z=3) → using the hydrogenic orbitals, the possible electron configuration are: 1s2 2p1, 1s2 2s1 or 1s3. The last proposal can be directly rejected because electrons are Fermions and they follow the Pauli exclusion principle: “No more than 2 electrons may occupy any given orbital. If 2 electrons occupy one orbital, then their spin must be paired (antiparallel: ↓↑).” The spin of the electron must be taken into account to understand the electronic configuration of atoms. A system of two paired electrons has a resultant spin of zero.
A. Pauli exclusion principle A system of Fermions (particles with half integral spin) is described by a Total wavefunction(x1, x2,..., xn) (where xi= ri, i.i is the spin coordinate of the electron i: or ), which is antisymmetric: when the labels of any two identical fermions are exchanged, the total wavefunction changes sign: (x1, x2,..., xn)= - (x2, x1,..., xn) In the orbital approximation: each electron is associated to a spinorbital that is the product between a spatial orbital and a spin function: (xi)= (ri) s(i) Let’s consider a system of 2 electrons that occupy the same spatial orbital (r). The total wave function of the system is (1,2)= [(1)(2)] S(1,2). Where the spatial part of the total wavefunction is (1)(2) and it is symmetric: (1)(2)= +(2)(1). Hence, in order to have the total wavefunction (1,2) antisymmetric, the total spinfunction S(1,2) must be antisymmetric: S(1,2)= -S(2,1).
What is this antisymmetric spinfunction S(1,2)? Let’s look at the different possibleassociation of 2 spins: - The two spins are “up” (↑↑) : S1(1,2)= (1)(2)→symmetric - The two spins are “down” (↓↓) : S2(1,2)= (1) (2)→symmetric - one “up” and one “down” (↑↓) …. but as the particle are indiscernible, we cannot distinguish between (1)(2) or (1)(2). Two linear combinations are then used in order to represent the situation where the spins are different: S3+(1,2)= 1/21/2 {(1)(2) + (1)(2)} →symmetric S4-(1,2)= 1/21/2 {(1)(2) - (1)(2)} →antisymmetric Only the spinfunction S4-(1,2) is antisymmetric and characterized by two different spins. That proofs the Pauli exclusion principle: 2 electrons in the same spatial orbital must be paired, i.e. must have different spins. The total wavefunction of a system of 2 electrons in the same orbital is: (1,2)= [(1)(2)] 1/21/2 {(1)(2) - (1)(2)} He: 2e- + nucleus (Z=2) → electron configuration: 1s2….and it is ↑↓ and not ↑↑
B. Penetration and shielding 31 2s 1s For hydrogenic atoms: 2s and 2p are degenerated For many-electron atoms: the 2s orbital lies lower in energy than the 2p orbitals…. Why? If the nucleus has a charge Z, the 2s electrons do not feel the potential created by Z, because the 1s electrons shield this nuclear charge. The 2s electrons feel an effective nuclear charge Zeff: Zeff= Z - is the shielding constant. This shielding constant is not the same for an electron in 2s and 2p, because these orbitals have different radial distributions (see Figure). A s orbital has a greater penetration through inner shells than a p orbital of the same shell. the 2s electrons feel a less shielded nuclear charge, i.e. more positively charged, than the 2p electrons. The 2s electrons are then more tightly bound to the nucleus. Usually the energies of subshells in a many-electron atom lie in the order: s<p<d<f Li: 3e- + nucleus (Z=2) → 1s2 2p1 is rejected, 1s2 2s1 is the correct configuration.
32 For heavy atoms, the rules for the energy order of the orbitals (s<p<d<f) fails. Example: the d subshells have 5 orbitals, a maximum of 10 electrons can be there…. These electrons are close to each other in space. For heavy atoms, the electron-electron repulsion energy is important and comparable to the energy difference between the 4s and 3d orbitals. That can lead to deviation of usual energy order of the orbitals (s<p<d<f)
C. Hund’s rule C: 6e- + nucleus (Z=6) → 1s2 2s2 2p2 …. The 2 last electrons in the 2p orbitals are the valence electrons. When adding electrons in the hydrogenic orbitals in order to estimate the electron configuration of atom (building-up principle), this rule has to be followed: “electrons occupy different orbitals of a given subshell before doubly occupying any one of them” This question is resolved by the empirical Hund’s rule: “An atom in its ground state adopts a configuration with the greatest number of unpaired electrons” It is then2px1 (↑) 2py1(↑). This is due to a quantum mechanical property:the spin correlation But is it 2px12py1 or 2px2? The 2 electrons repel each other by Coulombic repulsion, they prefer to be spatially far from each other. The configuration 2px2 is less stable than when the 2 electrons are on two different spatial orbitals: 2px12py1 is favorable. But is it 2px1 (↓) 2py1(↑) or 2px1 (↑) 2py1(↑)?
Let’s consider the situation where 2 electrons are in different orbitals a(1) and b(2) (like for C: 2px, 2py). The spatial part of the total wavefunction (1,2) cannot be the simple product: a(1)b(2), because it suggests that we know which electron is in which orbitals, whereas we cannot keep track of electrons. The correct description is a linear combination, either of the two following wavefunctions: +(1,2)= 1/21/2 {a(1)b(2) + b(1)a(2)} →symmetric -(1,2)= 1/21/2 {a(1)b(2) - b(1)a(2)} →antisymmetric In order to have an antisymmetric total wave function (1,2), +(1,2) should be multiplied by an antisymmetric spinfunction S(1,2) and -(1,2) by a symmetric spinfunction. S1(1,2)= +(1,2)S4-(1,2)= 1/2 {a(1)b(2) + b(1)a(2)}{(1)(2) - (1)(2)} T2(1,2)= -(1,2)S1(1,2)= 1/21/2{a(1)b(2) - b(1)a(2)}{(1)(2)} T3 (1,2)= -(1,2)S2(1,2)= 1/21/2{a(1)b(2) - b(1)a(2)}{(1) (2)} T4 (1,2)= -(1,2)S3+(1,2)= 1/2 {a(1)b(2) - b(1)a(2)} {(1)(2)+ (1)(2)} Antisymmetric spinfunction: Singlet Symmetric spinfunction: Triplet
What happens if the electron 1 approaches the electron 2: r1=r2 ? (↑↓) +(r1,r2=r1)= 1/21/2 {a(r1)b(r1) + b(r1)a(r1)} ≠ 0 ((1,2)= +(1,2)Santisym) (↑↑) -(r1,r2=r1)= 1/21/2 {a(r1)b(r1) - b(r1)a(r1)} = 0 ((1,2)= -(1,2)Ssym) When the two electrons are close to each other, the total wavefunction with an antisymmetric spinfunction, S1(r1,r2=r1) ≠0 :two electrons with different spin (antiparallel) have a certain probability to be at the same position. The electrostatic repulsion is increased:the system in itssinglet state is destabilized For two electrons having a symmetric spinfunction, the total wavefunction is zero: T2(r1,r2=r1)= T3(r1,r2=r1)= T4(r1,r2=r1)=0 : when 2 electrons in 2 different orbitals have the same spin(parallel), they are repelled to each other thanks to their spin. that prevents an additional electrostatic repulsion. The Triplet state (2 spins parallel) is more stable than the singlet state (2 spins antiparallel): this is the explanation for Hund’s rule. This quantum effect is called thespin correlation. The carbon atom has a valence configuration: 2px1 (↑) 2py1(↑) and not 2px1 (↑) 2py1(↓) .
3.2.2 Singlet and triplet states Triplet State : Symmetric spinfunction T2(1,2)= 1/21/2{a(1)b(2) - b(1)a(2)}{(1)(2)} T3 (1,2)= 1/21/2{a(1)b(2) - b(1)a(2)}{(1) (2)} T4 (1,2)= 1/2 {a(1)b(2) - b(1)a(2)} {(1)(2) + (1)(2)} When two electrons have “parallel” spins, they have a nonzero total spin angular momentum. Spin Multiplicity g= number of states having the same energy: g= 2S+1. Here, MS=1/2+1/2=1, there are 3 Triplet states with the same energy. Singlet State: Antisymmetric spinfunction S1(1,2)= 1/2 {a(1)b(2) + b(1)a(2)}{(1)(2) - (1)(2)} When two electrons have “antiparallel” spins, they have a zero total spin angular momentum. g= 2S+1. Here, S=1/2-1/2=0, there is only one Singlet state.
Spectrum of the He atom: Z=2, 2 s electrons. During the excitation, only one electron is excited. When the atom comes back to a lower energy state, two types of transitions are observed: Between triplet states: T → T, Between singlet states: S → S. But not: T → S or S → T The probability for a transition is given by the transition dipole momentfibetween an initial state i and a final state f : Both states of the atom are characterized by a spatial function and a spinfunction S. Spin selection rule If the initial and final states have both a spinfunction of the same symmetry, the transition dipole moment is non-zero: the transition is allowed. If the two states have different spinfunction symmetries, the transition is forbidden.