Download
1 / 1
10 likes | 17 Views
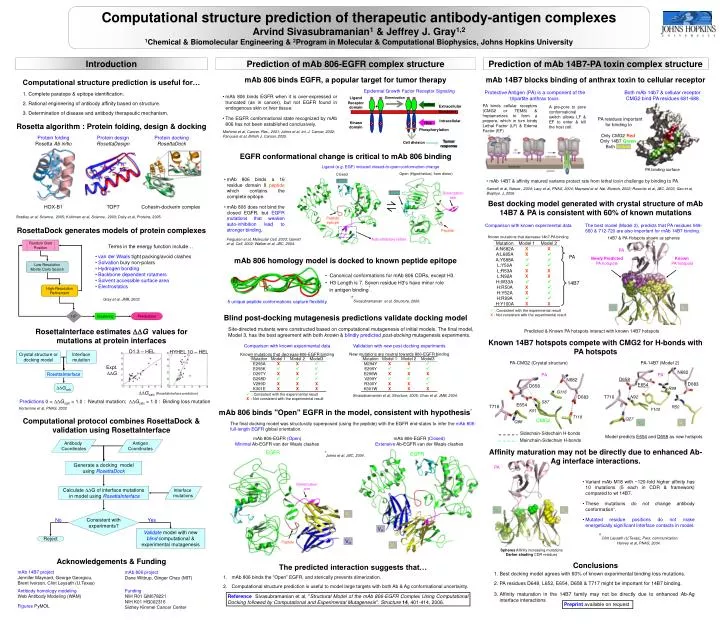
*. Sivasubramanian et al, Structure, 2006. Sidechain-Sidechain H-bonds. Mainchain-Sidechain H-bonds. *. Clint Leysath (U.Texas), Pers. communication. Computational structure prediction of therapeutic antibody-antigen complexes Arvind Sivasubramanian 1 & Jeffrey J. Gray 1,2

E N D
* Sivasubramanian et al, Structure, 2006. Sidechain-Sidechain H-bonds Mainchain-Sidechain H-bonds * Clint Leysath (U.Texas), Pers. communication. Computational structure prediction of therapeutic antibody-antigen complexes Arvind Sivasubramanian1 & Jeffrey J. Gray1,2 1Chemical & Biomolecular Engineering & 2Program in Molecular & Computational Biophysics, Johns Hopkins University Prediction of mAb 806-EGFR complex structure Prediction of mAb 14B7-PA toxin complex structure Introduction mAb 806 binds EGFR, a popular target for tumor therapy mAb 14B7 blocks binding of anthrax toxin to cellular receptor Computational structure prediction is useful for… Epidermal Growth Factor Receptor Signaling Protective Antigen (PA) is a component of the tripartite anthrax toxin. Both mAb 14b7 & cellular receptor CMG2 bind PA residues 681-688 • Complete paratope & epitope identification. • Rational engineering of antibody affinity based on structure. • Determination of disease and antibody therapeutic mechanism. • mAb 806 binds EGFR when it is over-expressed or truncated (as in cancer), but not EGFR found in endogenous skin or liver tissue. • The EGFR conformational state recognized by mAb 806 has not been established conclusively. Ligand Dimerization Receptor domain mAbs + PA binds cellular receptors (CMG2 or TEM8) & heptamerizes to form a prepore, which in turn binds Lethal Factor (LF) & Edema Factor (EF) Extracellular A pre-pore to pore conformational switch allows LF & EF to enter & kill the host cell. Membrane PA residues important for binding to Only CMG2 Red Only 14B7 Green Both Yellow Intracellular Kinase domain TKIs Rosetta algorithm : Protein folding, design & docking K K K K Phosphorylation Mishima et al, Cancer. Res., 2001; Johns et al, Int. J. Cancer, 2002; Panousis et al, British J. Cancer, 2005. Protein folding Rosetta Ab initio Protein design RosettaDesign Protein docking RosettaDock Tumor response Cell division Yellow EGFR conformational change is critical to mAb 806 binding Ligand (e.g. EGF) induced closed-to-open conformation change PA binding surface Open (Hypothetical, from dimer) Closed • mAb 806 binds a 16 residue domain II peptide which contains the complete epitope. • mAb 806 does not bind the closed EGFR, but EGFR mutations that weaken auto-inhibition lead to stronger binding. • mAb 14B7 & affinity matured variants protect rats from lethal toxin challenge by binding to PA. Ligand Santelli et al, Nature , 2004; Lacy et al, PNAS, 2004; Maynard et al, Nat. Biotech, 2002; Rosovitz et al, JBC, 2003, Gao et al, Biophys. J, 2006. Dimerization arm Ligand ⇌ Best docking model generated with crystal structure of mAb 14B7 & PA is consistent with 60% of known mutations HOX-B1 TOP7 Cohesin-dockerin complex Bradley et al, Science, 2005; Kuhlman et al, Science, 2003; Daily et al, Proteins, 2005. Peptide epitope Comparison with known experimental data The best model (Model 2), predicts that PA residues 648-660 & 712-720 are also important for mAb 14B7 binding. RosettaDock generates models of protein complexes Peptide Known mutations that decrease 14b7-PA binding 14B7 & PA Hotspots shown as spheres Ferguson et al, Molecular Cell, 2003; Garrett et al, Cell, 2002; Walker et al, JBC, 2004. Auto-inhibitory tether Random Start Position Terms in the energy function include… PA • van der Waals tight packing/avoid clashes • Solvation bury non-polars • Hydrogen bonding • Backbone dependent rotamers • Solvent accessible surface area • Electrostatics mAb 806 homology model is docked to known peptide epitope PA Newly Predicted PA hotspots Known PA hotspots Low-Resolution Monte Carlo Search • Canonical conformations for mAb 806 CDRs, except H3. • H3 Length is 7. Seven residue H3's have minor role in antigen binding*. 14B7 High-Resolution Refinement Gray et al, JMB, 2003. 5 unique peptide conformations capture flexibility. VH VL : Consistent with the experimental result X : Not consistent with the experimental result 105 Predictions Blind post-docking mutagenesis predictions validate docking model Clustering Site-directed mutants were constructed based on computational mutagenesis of initial models. The final model, Model 3, has the best agreement with both known & blindly predictedpost-docking mutagenesis experiments. RosettaInterface estimates G values for mutations at protein interfaces Predicted & Known PA hotspots interact with known 14B7 hotspots Known 14B7 hotspots compete with CMG2 for H-bonds with PA hotspots Comparison with known experimental data Validation with new post-docking experiments D1.3 – HEL HYHEL 10 – HEL Crystal structure or docking model Interface mutation New mutations are neutral towards 806-EGFR binding Known mutations that decrease 806-EGFR binding PA-CMG2 (Crystal structure) PA-14B7 (Model 2) Expt. G N682 RosettaInterface PA PA D658 N682 D683 E654 D658 Gcalc R99 G116 Gcalc(RosettaInterface prediction) : Consistent with the experimental result X: Not consistent with the experimental result Sivasubramanian et al, Structure, 2006; Chao et al, JMB, 2004. D683 T716 N92 Predictions 0 < Gcalc < 1.0 : Neutral mutation; Gcalc > 1.0 : Binding loss mutation S87 E654 T716 R50 Kortemme et al, PNAS, 2002 Y100 mAb 806 binds "Open" EGFR in the model, consistent with hypothesis* K51 T118 Q27 Computational protocol combines RosettaDock & validation using RosettaInterface CMG2 Q88 VH VL The final docking model was structurally superposed (using the peptide) with the EGFR end-states to infer the mAb 806-full-length EGFR global orientation. Model predicts E654 and D658 as new hotspots mAb 806-EGFR (Open) Minimal Ab-EGFR van der Waals clashes mAb 806-EGFR (Closed) Extensive Ab-EGFR van der Waals clashes Antibody Coordinates Antigen Coordinates Affinity maturation may not be directly due to enhanced Ab-Ag interface interactions. EGFR EGFR *Johns et al, JBC, 2004. Generate a docking model using RosettaDock PA • Variant mAb M18 with ~120-fold higher affinity has 10 mutations (5 each in CDR & framework) compared to wt 14B7. • These mutations do not change antibody conformation*. • Mutated residue positions do not make energetically significant interface contacts in model. Dimerization arm Calculate G of interface mutations in model using RosettaInterface Interface mutations VH VL Consistent with experiments? VL VL No Yes VH Validate model with new blindcomputational & experimental mutagenesis Reject VH VH Peptide Harvey et al, PNAS, 2004. VL Spheres Affinity increasing mutations Darker shading CDR residues Acknowledgements & Funding Conclusions The predicted interaction suggests that… • Best docking model agrees with 60% of known experimental binding loss mutations. • PA residues D648, L652, E654, D658 & T717 might be important for 14B7 binding. • Affinity maturation in the 14B7 family may not be directly due to enhanced Ab-Ag interface interactions • mAb 806 binds the “Open” EGFR, and sterically prevents dimerization. • Computational structure prediction is useful to model large targets with both Ab & Ag conformational uncertainty. mAb 14B7 project Jennifer Maynard, George Georgiou, Brent Iverson, Clint Leysath (U.Texas) mAb 806 project Dane Wittrup, Ginger Chao (MIT) Antibody homology modeling Web Antibody Modeling (WAM) Funding NIH R01 GM078221 NIH K01 HG002316 Sidney Kimmel Cancer Center Reference Sivasubramanian et al, "Structural Model of the mAb 806-EGFR Complex Using Computational Docking followed by Computational and Experimental Mutagenesis". Structure14, 401-414, 2006. Preprint available on request Figures PyMOL