Download

1 / 43
440 likes | 581 Views
Blood Clotting. April 21, 2005 . Blood clotting system. Precise control of the blood-clotting system is essential for maintenance of the circulation in all higher animals. Deficient function of this system can lead to fatal bleeding following even a minor injury.

E N D
Blood Clotting April 21, 2005
Blood clotting system • Precise control of the blood-clotting system is essential for maintenance of the circulation in all higher animals. • Deficient function of this system can lead to fatal bleeding following even a minor injury. • Overactivity of this system can produce unwanted blood clots, resulting in blockages to critical blood vessels, as occurs in such diseases as heart attack and stroke.
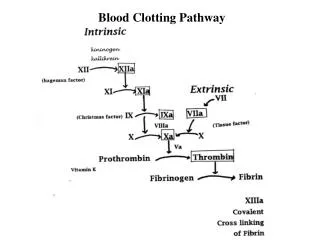
Hemostasis in Physiological Conditions • Under homeostatic conditions, the body is maintained in a finely tuned balance of coagulation and fibrinolysis.. The activation of the coagulation cascade yields thrombin that converts fibrinogen to fibrin; the stable fibrin clot being the final product of hemostasis. • The fibrinolytic system then functions to break down fibrinogen and fibrin. Activation of the fibrinolytic system generates plasmin (in the presence of thrombin), which is responsible for the lysis of fibrin clots. • The breakdown of fibrinogen and fibrin results in polypeptides called FDPs or fibrin split products (FSPs). • In a state of homeostasis, the presence of thrombin is critical, as it is the central proteolytic enzyme of coagulation and is also necessary for the breakdown of clots, or fibrinolysis.
DIC • DIC is a state of hypercoagulation that occurs in a variety of disease states. • DIC represents an inappropriate overstimulation of normal coagulation in which thrombosis and hemorrhage occur simultaneously. • Hypercoagulation occurs initially in the process of DIC; multiple small clots are formed in the microcirculation of various organs. This process is followed by fibrinolysis, in which there is consumption of clots and clotting factors, resulting in bleeding. • Finally, the body is unable to respond to vascular or tissue injury through stable clot formation, and hemorrhage occurs. The hemorrhage associated with DIC can be profound, but it is the diffuse thrombosis (both microvascular and large vessel involvement) that leads to the irreversible morbidity and mortality associated with DIC. It is the thrombosis that leads to ischemia, impairment of blood flow, and end organ damage.
DIC • DIC can be acute or chronic in nature. • Chronic DIC is generally seen in the cancer population and is demonstrated as localized thrombotic events (eg, deep vein thromboses). Chronic DIC is defined as a state of intravascular coagulation, and only minor imbalances in hemostasis exist. • The acute form of DIC is considered an extreme expression of the intravascular coagulation process with a complete breakdown of the normal homeostatic boundaries. DIC is associated with a poor prognosis and a high mortality rate.
DIC-pathophysiology • In DIC, the processes of coagulation and fibrinolysis lose control, and the result is widespread clotting with resultant bleeding. • Regardless of the triggering event of DIC, once initiated, the pathophysiology of DIC is similar in all conditions. • One critical mediator of DIC is the release of a transmembrane glycoprotein called tissue factor (TF). TF is present on the surface of many cell types (including endothelial cells, macrophages, and monocytes) and is not normally in contact with the general circulation, but is exposed to the circulation after vascular damage. For example, TF is released in response to exposure to cytokines (particularly interleukin), tumor necrosis factor, and endotoxin. This plays a major role in the development of DIC in septic conditions. TF is also abundant in tissues of the lungs, brain, and placenta. This helps to explain why DIC readily develops in patients with extensive trauma. • TF binds with coagulation factors that then trigger both the intrinsic and the extrinsic pathways of coagulation.
DIC • Excess circulating thrombin results from the excess activation of the coagulation cascade. The excess thrombin cleaves fibrinogen, which ultimately leaves behind multiple fibrin clots in the circulation. • These excess clots trap platelets to become larger clots, which leads to microvascular and macrovascular thrombosis. This lodging of clots in the microcirculation, in the large vessels, and in the organs is what leads to the ischemia, impaired organ perfusion, and end-organ damage that occurs with DIC.
DIC • Simultaneously, excess circulating thrombin assists in the conversion of plasminogen to plasmin, resulting in fibrinolysis. The breakdown of clots results in excess amounts of FDPs, which have powerful anticoagulant properties, contributing to hemorrhage. • The excess plasmin also activates the complement and kinin systems. Activation of these systems leads to many of the clinical symptoms that patients experiencing DIC exhibit, such as shock, hypotension, and increased vascular permeability.
DIC • Coagulation inhibitors are also consumed in this process. Decreased inhibitor levels will permit more clotting so that a feedback system develops in which increased clotting leads to more clotting. • Thrombocytopenia occurs because of the entrapment of platelets. Clotting factors are consumed in the development of multiple clots, which contributes to the bleeding seen with DIC.
DIC • The fibrinolytic system, the body's mechanism for breaking down blood clots, is delicately balanced with the system that forms blood clots. Overactivity of either system results in uncontrolled bleeding or uncontrolled blood clotting. Plasminogen activators are the proteins that turn on the fibrinolytic system. Their activity is controlled by several regulator proteins, including plasminogen activator inhibitor-1 (PAI1) and PAI2.
Hypocoagulation • Inherited states • Acquired states
Vitamin K Deficiency • Insufficient amounts of vitamin K in diet • Inadequate synthesis by gastrointestinal bacteria • Abnormal absorbtion from small intestine • Drugs (antivitamins K-coumadin)
Clinical Picture of Vitamin K Deficiency • Bleeding in: • Hospitalized patients on intravenous fluids, with broad-spectrum antibiotics that sterilize the gut • Newborn (premature) infants with immature liver function • Abnormalities in fat absorbption • Deficiency in bile salts
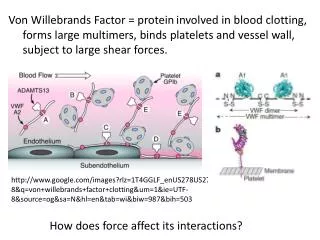
von Willebrand factor • The blood-clotting protein (VWF) functions as the critical initial bridge connecting blood platelets to the wall of injured blood vessels, thereby helping to control bleeding. • VWF also serves as the carrier for factor VIII, the substance missing in patients with hemophilia. Abnormalities in VWF result in von Willebrand disease (VWD), the most common human inherited bleeding disorder.
von Willebrand Factor-genetics • The molecular basis for the most common variant (type 1) still remains largely a mystery. • In some patients, a mutation inactivating one copy of the VWF gene reduces plasma VWF, leading to bleeding; in others, the same change does not result in a significant problem. • It is now clear that wide variations in disease among individuals with the same or similar defects in the VWF gene are due to the action of one or more additional "modifier" genes. Such modifier genes are also likely to contribute to the wide variation in VWF levels observed among normal individuals. Elevated levels of VWF produced in this way may result in an increased risk of blood clotting, in contrast to the bleeding tendency associated with low VWF.
Trombotic thrombocytopenic purpura (TTP) • A deficiency of a protein that normally partially breaks down VWF in the blood as the cause of the often catastrophic blood-clotting disease (TTP) was identified. • Mutations in ADAMTS13, the gene for this protein, in nearly all patients with the inherited form of TTP were found. • Identification of this gene provides new tools for improved diagnosis and should lead to the production of recombinant ADAMTS13 for more effective treatment of TTP.
Defects Responsible for Hypercoagulability-Inherited • Activated protein C resistance (factor V Leiden) • Protein S deficiency • Protein C deficiency • Antithrombin deficiency • Hyperhomocysteinemia • Prothrombin 20210A allele • Dysplasminogenemia • High plasminogen activator inhibitor • Dysfibrinogenemia • Elevated factor VIII
Defects Responsible for Hypercoagulability-Acquired • Antiphospholipid syndrome • Hyperhomocysteinemia • Miscellaneous • Thrombocythemia • Dysproteinemia • Heparin-induced thrombocytopenia • Estrogens • Birth control pills • Hormone replacement therapy
Defects Responsible for Hypercoagulability-Noncoagulant factors • Malignancy • Pregnancy • Bed rest • Surgery • Trauma
Coagulation Factor V • Coagulation factor V is a central regulator in the early phases of blood clot formation. Genetic deficiency of factor V results in a rare bleeding disorder called parahemophilia. • A subtle change in the factor V gene that increases the function of this protein, called factor V Leiden, is an important cause of an abnormal increase in blood clot formation. • Factor V Leiden is remarkably common (present in 2–7 percent of the population) and may contribute to up to 50 percent of hospital admissions for blood clot–related illnesses. • 10 percent of humans with factor V Leiden will develop a serious blood clot during their lifetime from the 90 percent who will remain asymptomatic.
Combined deficiency of coagulation factor V and coagulation factor VIII • Combined deficiency of coagulation factor V and coagulation factor VIII is another inherited bleeding disease. • The molecular basis for this disorder as deficiency of the cellular protein LMAN1 (also known as ERGIC53) was identified. • Though LMAN1 gene mutations in many combined deficiency patients were found, the cause of this disorder in approximately 30 percent of these individuals remained unexplained. In these patients, mutations in another gene, termed MCFD2 was found. • A complex of MCFD2 and LMAN1 appears to serve as a carrier for a subset of proteins, including factors V and VIII, that are destined for export from the cell. These findings provide the first example of such a specific transport pathway within the cells of higher organisms.
Antithrombin III • In the final phase of clot formation, thrombin converts fibrinogen to fibrin. Antithrombin (formerly referred to as antithrombin III), named for its action on thrombin, also inhibits the serine proteases of IXa, Xa, XIa, and XIIa. • Deficiency of antithrombin may be caused by decreased levels or by dysfunctional protein. The "anticoagulant" action of heparin requires the presence of antithrombin; thus, a clinical clue to diagnosis of antithrombin deficiency may be anticoagulation refractoriness to heparin.
Heparin-Induced Hypercoagulability • Heparin associated thrombocytopenia is often seen, but "heparin-induced hypercoagulability" is infrequent. • IgG antibodies form against platelet-heparin complexes that are sequestered on platelets at platelet Fc receptors and on endothelial cells where they may cause serious vascular occlusive disease and thrombocytopenia. • Warfarin accelerates this phenomenon by further decreasing proteins of the protein C pathways, thereby enhancing hypercoagulability.The treatment of heparin-induced hypercoagulability, including purpura fulminans, requires immediate discontinuance of heparin administration. • Low molecular weight heparin (LMWH) is risky because of potential cross reactivity with heparin antibodies.
Activated Protein C Resistance • The pathophysiologic mechanism of resistance of APC is related to an inherited abnormality of factor V. Investigations have identified a genetic point mutation on chromosome 1 that encodes glutamine at the 506 site rather than arginine (ARG 506 GLN). This modification is responsible for the production of an aberrant factor V that is resistant to the proteolytic destruction by APC. • Aberrant factor V was originally described in Leiden (Holland) and is more commonly referred to as factor V Leiden. When normal factor V is digested at the arginine 506 site, two other sites cooperate; 70% of the destruction of factor Va occurs at arginine 306, and 30% occurs at arginine 679. Protein S cooperatively acts upon arginine 306; therefore, even in the presence of factor V Leiden, there is a lesser degree of thrombosis when protein S is present. Conversely, when protein S deficiency coexists with factor V Leiden, thrombotic events are more prevalent. Except for other rare genetic abnormalities (factor V Hong Kong and factor V Cambridge, factor V Leiden is synonymous with the term APC resistance.
* From Olds et al.[26] APC = Activated protein C. Data on prothrombin 20210A are approximated from multiple sources.[23,64-66] Table 3. Charges for Hypercoagulability Tests* * Data obtained from nationally recognized reference laboratory. APC = Activated protein C. Testing is often more expensive when ordered on hospitalized patients. Table 4. Factors Responsible for Altered Coagulation Values* • * Data from Adcock et al.[23] • Malignancy can represent one or more of these factors, such as inflammation, liver disease, surgery, disseminated intravascular coagulation. • References • Bauer KA: Hypercoagulable states. Hematology: Basic Principles and Practice. Hoffman R, Benz EJ Jr, Shattil SJ, et al (eds). New York, Churchill Livingstone, 1995, pp 1781-1795 • Allaart CF, Briet E: Familial venous thrombophilia. Haemostasis and Thrombosis. Bloom AL, Forbes CD, Thomas DP, et al (eds). London, Churchill Livingstone, 1994, p 1349 • Svensson PJ, Dahlback B: Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med 1994; 330:517-522 • Koster T, Rosendaal FR, de Ronde H, et al: Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet 1993; 342:1503-1506 • Griffin JH, Evatt B, Wideman C, et al: Anticoagulant protein C pathway defective in majority of thrombophilic patients. Blood 1993; 82:1989-1993 • Harris JM, Abramson N: Evaluation of recurrent thrombosis and hypercoagulability. Am Fam Physician 1997; 56:1591-1596,1601 • Gandrille S, Greengard JS, Alhenc-Gelas M, et al: Incidence of activated protein C resistance caused by the ARG 506 GLN mutation in factor V in 113 unrelated symptomatic protein C-deficient patients. The French Network on the behalf of INSERM. Blood 1995; 86:219-224 • Bertina RM, Koeleman BP, Koster T, et al: Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369:64 • Griffin JH, Heeb MJ, Kojima Y, et al: Activated protein C resistance: molecular mechanisms. Thromb Haemost 1995; 74:444-448 • Zivelin A, Griffin JH, Xu X, et al: A single genetic origin for a common caucasian risk factor for venous thrombosis. Blood 1997; 89:397-402 • Rees DC, Cox M, Clegg JB: World distribution of factor V Leiden. Lancet 1995; 346:1133-1134 • Cox MJ, Rees DC, Martinson JJ, et al: Evidence for a single origin of factor V Leiden. Br J Haematol 1996; 92:1022-1025 • Middeldorp S, Henkens CM, Koopman MM, et al: The incidence of venous thromboembolism in family members of patients with factor V Leiden mutation and venous thrombosis. Ann Intern Med 1998; 128:15-20 • Hellgren M, Svensson PJ, Dahlback B: Resistance to activated protein C as a basis for venous thromboembolism associated with pregnancy and oral contraceptives. Am J Obstet Gynecol 1995; 173:210-213 • Brenner B, Lanir N, Thaler I: HELLP syndrome associated with factor V R506Q mutation. Br J Haematol 1996; 92:999-1001 • Cumming AM, Tait RC, Fildes S, et al: Diagnosis of APC Resistance during pregnancy. Br J Haematol 1996; 92:1026-1029 • Cumming AM, Tait RC, Fildes S, et al: Development of resistance to activated protein C during pregnancy. Br J Haematol 1995; 90:725-727 • Mathonnet F, de Mazancourt P, Bastenaire B, et al: Activated protein C sensitivity ratio in pregnant women at delivery. Br J Haematol 1996; 92:244-246 • Olivieri O, Friso S, Manzato F, et al: Resistance to activated protein C in healthy women taking oral contraceptives. Br J Haematol 1995; 91:465-470 • Heinrich J, Budde T, Assmann G: Mutation in the factor V gene and the risk of myocardial infarction (Letter). N Engl J Med 1995; 333:881 • Ridker PM, Hennekens CH, Lindpaintner K, et al: Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995; 332:912-917 • Koeleman BP, Reitsma PH, Allaart CF, et al: Activated protein C resistance as an additional risk factor for thrombosis in protein C-deficient families. Blood 1994; 84:1031-1035 • Poort SR, Rosendaal FR, Reitsma PH, et al: A common genetic variation in the 3'-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 1996; 88:3698-3703 • Ames PR, Tommasino C, D'Andrea G, et al: Thrombophilic genotypes in subjects with idiopathic antiphospholipid antibodies -- prevalence and significance. Thromb Haemost 1998; 79:46-49 • Kamphuisen PW, Lensen R, Houwing-Duistermaat JJ, et al: Heritability of elevated factor VIII antigen levels in factor V Leiden families with thrombophilia. Br J Haematol 2000; 109:519-522 • Olds RJ, Fitches AC, Geary CP: The multigenic basis for venous thrombosis. Br J Haematol 2000; 109:508-511 • Mandel H, Brenner B, Berant M, et al: Coexistence of hereditary homocystinuria and factor V Leiden -- effect on thrombosis. N Engl J Med 1996; 334:763-768 • den Heijer M, Koster T, Blom HJ, et al: Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996; 334:759-762 • Griffin JH, Evatt B, Zimmerman TS, et al: Deficiency of protein C in congenital thrombotic disease. J Clin Invest 1981; 68:1370-1373 • Mahasandana C, Suvatte V, Marlar RA, et al: Neonatal purpura fulminans associated with homozygous protein S deficiency. Lancet 1990; 1:61-62 • Marciniak E, Wilson HD, Marlar RA: Neonatal purpura fulminans: a genetic disorder related to the absence of protein C in blood. Blood 1985; 65:15-20 • Marlar RA, Montgomery RR, Broekmans AW: Diagnosis and treatment of homozygous protein C deficiency. report of the Working Party on Homozygous Protein C Deficiency of the Subcommittee on Protein C and Protein S, International Committee on Thrombosis and Haemostatis. J Pediatr 1989; 114:528-534 • Lane DA, Caso R: Antithrombin: structure, genomic organization, function and inherited deficiency. The Molecular Biology of Coagulation. Bailliere's Clinical Haematology. Tuddenham EGD (ed). London, Bailliere Tindall, 1989, p 961 • Abildgaard U: Antithrombin and related inhibitors of coagulation. Recent Advances in Blood Coagulation. Poller L (ed). Edinburgh, Churchill Livingstone, 1981, p 146 • Fermo I, Vigano D'Angelo S, Paroni R, et al: Prevalence of moderate hyperhomocysteinemia in patients with early-onset venous and arterial occlusive disease. Ann Intern Med 1995; 123:747-753 • D'Angelo A, Selhub J: Homocysteine and thrombotic disease. Blood 1997; 90:1-11 • Mayer EL, Jacobsen DW, Robinson K: Homocysteine and coronary atherosclerosis. J Am Coll Cardiol 1996; 27:517-527 • Zivelin A, Rosenberg N, Faier S, et al: A single genetic origin for the common prothrombotic G20210A polymorphism in the prothrombin gene. Blood 1998; 92:1119-1124 • Makris M, Preston FE, Beauchamp NJ, et al: Co-inheritance of the 20210A allele of the prothrombin gene increases the risk of thrombosis in subjects with familial thrombophilia. Thromb Haemost 1997; 78:1426-1429 • Koster T, Blann AD, Briet E, et al: Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995; 345:152-155 • Nachman RL, Silverstein R: Hypercoagulable states. Ann Intern Med 1993; 119:819-827 • Kyrle PA, Minar E, Hirschl M, et al: High plasma levels of factor VIII and the risk of recurrent thromboembolism. N Engl J Med 2000; 343:457-462 • Ginsberg JS, Wells PS, Brill-Edwards P, et al: Antiphospholipid antibodies and venous thromboembolism. Blood 1995; 86:3685-3691 • Lockshin MD: Answers to the antiphospholipid-antibody syndrome? (editorial; comment). N Engl J Med 1995; 332:1025-1027 • Santoro SA: Antiphospholipid antibodies and thrombotic predisposition: underlying pathogenetic mechanisms (editorial; comment). Blood 1994; 83:2389-2391 • Bick RL: The antiphospholipid-thrombosis syndromes. fact, fiction, confusion, and controversy (Editorial). Am J Clin Pathol 1993; 100:477-480 • Ginsberg JS, Brill-Edwards P, Johnston M, et al: Relationship of antiphospholipid antibodies to pregnancy loss in patients with systemic lupus erythematosus: a cross-sectional study. Blood 1992; 80:975-980 • Galli M, Finazzi G, Barbui T: Annotation: thrombocytopenia in the antiphospholipid syndrome. Br J Haematol 1996; 93:1-5 • Lemmers NW, Gels ME, Sleijfer DT, et al: Complications of venous access ports in 132 patients with disseminated testicular cancer treated with polychemotherapy. J Clin Oncol 1996; 14:2916-2922 • Eastridge BJ, Lefor AT: Complications of indwelling venous access devices in cancer patients. J Clin Oncol 1995; 13:233-238 • Nowak-Gottl U, Wermes C, Junker R, et al: Prospective evaluation of the thrombotic risk in children with acute lymphoblastic leukemia carrying the MTHFR TT 677 genotype, the prothrombin G20210A variant, and further prothrombotic risk factors. Blood 1999; 93:1595-1599 • Boraks P, Seale J, Price J, et al: Prevention of central venous catheter associated thrombosis using minidose warfarin in patients with haematological malignancies. Br J Haematol 1998; 101:483-486 • Hutten BA, Prins MH, Gent M, et al: Incidence of recurrent thromboembolic and bleeding complications among patients with venous thromboembolism in relation to both malignancy and achieved international normalized ratio: a retrospective analysis. J Clin Oncol 2000; 18:3078-3083 • Warkentin TE, Elavathil LJ, Hayward CP, et al: The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann Intern Med 1997; 127:804-812 • Argatroban approved for heparin-induced thrombocytopenia (News). Am J Health Syst Pharm 2000; 57:1650 • Adcock DM, Fink L, Marlar RA: A laboratory approach to the evaluation of hereditary hypercoagulability. Am J Clin Pathol 1997; 108:434-449 • Henkens CM, Bom VJ, Van der Schaaf W, et al: Plasma levels of protein S, protein C, and factor X: effects of sex, hormonal state and age. Thromb Haemost 1995; 74:1271-1275 • Henkens CM, Bom VJ, Seinen AJ, et al: Sensitivity to activated protein C; influence of oral contraceptives and sex. Thromb Haemost 1995; 73:402-404 • Simonneau G, Sors H, Charbonnier B, et al: A comparison of low-molecular-weight heparin with unfractionated heparin for acute pulmonary embolism. the THESEE Study Group. Tinzaparine ou Heparine Standard: Evaluations dans l'Embolie Pulmonaire. N Engl J Med 1997; 337:663-669 • Epstein DJ, Bergum PW, Bajaj SP, et al: Radioimmunoassays for protein C and factor X. plasma antigen levels in abnormal hemostatic states. Am J Clin Pathol 1984; 82:573-581 • De Stefano V, Mastrangelo S, Schwarz HP, et al: Replacement therapy with a purified protein C concentrate during initiation of oral anticoagulation in severe protein C congenital deficiency. Thromb Haemost 1993; 70:247-249 • Gabriel DA: The use of antithrombin III in the treatment of disseminated intrasvascular coagulation. Semin Hematol 1994; 31:60-64 • Khamashta MA, Cuadrado MJ, Mujic F, et al: The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 1995; 332:993-997 • Hillarp A, Zoller B, Svensson PJ, et al: The 20210 A allele of the prothrombin gene is a common risk factor among Swedish outpatients with verified deep venous thrombosis. Thromb Haemost 1997; 78:990-992 • Brown K, Luddington R, Williamson D, et al: Risk of venous thromboembolism associated with a G to A transition at position 20210 in the 3'-untranslated region of the prothrombin gene. Br J Haematol 1997; 98:907-909 • Cumming AM, Keeney S, Salden A, et al: The prothrombin gene G20210A variant: prevalence in a U.K. anticoagulant clinic population. Br J Haematol 1997; 98:353-355 • Funding InformationSupported by the Baptist Regional Cancer Institute Research Foundation. • Reprint AddressReprint requests to Neil Abramson, MD, Baptist Regional Cancer Institute, Department of Education and Research, Jacksonville, FL 32207.
Protein C and Protein S Deficiencies • Protein C and protein S are vitamin K-dependent factors that are synthesized in the liver. • Protein C originates on chromosome 2 and protein S on chromosome 3. • Deficiencies of these proteins have been considered autosomal dominant defects, though recent analyses suggest that the defects may be recessive but with a high frequency of concomitant defects of other coagulation proteins. • Two types of protein defects cause protein C or protein S deficiency: deficiency of protein content (antigen) or the presence of dysfunctional protein. Protein S deficiency occurs at a slightly greater frequency than does protein C deficiency. Heterozygote protein C and protein S abnormalities cause hypercoagulability; in rare instances, homozygote protein C or homozygote protein S deficiency can result in a life-threatening coagulopathy of neonates (purpura fulminans).
Prothrombin 20210A • Frequency of this abnormality varies from 0.7% to 6.0% among whites, with rare appearances among Africans and Asians, suggesting that the defect may have also appeared after the divergent migrations of the populations. • The combination of prothrombin 20210A with other defects such as factor V Leiden, protein S deficiency, protein C deficiency, or antithrombin deficiency has been reported. • The mechanism by which prothrombin 20210A allele is responsible for hypercoagulability is uncertain.
Other Inherited Disorders • Other inheritable hypercoagulable diseases such as increased levels of factor VIII have been recently reported. • Dysplasminogenemia,hypoplasminogenemia, decreased release of tissue plasminogen activator, and increased concentrations of plasminogen activator inhibitor may occur rarely but are not well established. • Dysfibrinogenemias are usually manifested as bleeding disorders because of defective fibrin formation, but thrombotic complications may occur when the defective fibrin is resistant to the lytic effects of plasmin. Recent descriptions of elevated levels in factor VIII suggest an etiologic mechanism for recurrent thromboembolic disease; factor VIII elevations may be increased by inherited and acquired factors.
Antiphospholipid Syndrome • The "lupus anticoagulant" is an acquired biologic abnormality characterized as an "anticoagulant" in vitro but associated with excessive clotting in vivo. • This abnormality, also referred to as the antiphospholipid syndrome, should be suspected in young persons with arterial disease such as myocardial infarctions and acute neurologic events (cerebrovascular accidents and transient ischemic attacks). • is seen in women with recurrent pregnancy loss or in patients with increased thrombosis especially in unusual locations such as retinal veins, cerebral vessels, and hepatic venous channels (Budd-Chiari syndrome). Patients with antiphospholipid syndrome may have mild thrombocytopenia as well.
Other Acquired Disorders • Malignancy, pregnancy, surgery, connective tissue diseases, lymphoproliferative diseases, myeloproliferative disorders, and dysproteinemias are recognized causes of hypercoagulability, but the mechanisms are unclear and may vary with each situation. • With malignancy, excessive clotting is allegedly related to thromboplastin-like effects produced by tumor cells or their products. Mucin-producing malignancy has a high association of thrombosis. Excessive clotting with malignancy may also be caused by concomitant infections, effects of chemotherapy, malnutrition and possible folate deficiency with its consequences on homocysteine, and prolonged bed rest. Venous access devices that are commonly used in cancer patients predispose to clotting.
Other Acquired Disorders • Cancer patients often receive low-dose warfarin (1 to 2 mg/day) to lessen the frequency of veno-occlusive disease. Cancer patients also are relatively resistant to anticoagulation and have more episodes of recurrent thrombosis. • Pregnancy associated clotting may relate to excess thromboplastin production; hypercoagulability is more frequent with pregnancy complications, such as abruptio placenta, amniotic fluid embolization, and retained dead fetus. Mechanisms of hypercoagulability with surgery are less clear but may relate to tissue trauma and/or the effects of bed rest. Previous clotting also predisposes to recurrence; some factors include clotting associated abnormalities in the anatomy of the vasculature and associated increased levels of coagulation factors as acute phase reactants.